

Challenges and Prospects in the Catalytic Conversion of Carbon Dioxide to Formaldehyde
Abstract
Formaldehyde (HCHO) is a crucial C1 building block for daily-life commodities in a wide range of industrial processes. Industrial production of HCHO today is based on energy- and cost-intensive gas-phase catalytic oxidation of methanol, which calls for exploring other and more sustainable ways of carrying out this process. Utilization of carbon dioxide (CO2) as precursor presents a promising strategy to simultaneously mitigate the carbon footprint and alleviate environmental issues. This Minireview summarizes recent progress in CO2-to-HCHO conversion using hydrogenation, hydroboration/hydrosilylation as well as photochemical, electrochemical, photoelectrochemical, and enzymatic approaches. The active species, reaction intermediates, and mechanistic pathways are discussed to deepen the understanding of HCHO selectivity issues. Finally, shortcomings and prospects of the various strategies for sustainable reduction of CO2 to HCHO are discussed.
1 Introduction
For decades formaldehyde (HCHO) has constituted an essential building block in the production of daily-life commodities such as biocides, disinfectants, preservatives, glues/adhesives, and resins.1, 2 Worldwide production of HCHO is estimated to be in excess of 45.6 million metric tons per year, with an ever growing demand. At present, the Formox Process is the main source of HCHO. It includes an energy-intensive three-step process line consisting of steam reforming natural gas (700–1100 °C) to syngas, followed by methanol (CH3OH) synthesis (200–300 °C) and partial oxidation/dehydrogenation of CH3OH (300–400 °C),3 resulting in both ecological and economic deficiencies. Thus, there is an urgent need to identify more sustainable processes for producing HCHO.
At the same time, excessive utilization of fossil fuels has led to rapid rise of the atmospheric carbon dioxide (CO2) level, triggering climate and environmental issues (e.g., green-house effect and sea-level rise) and energy crises. To mitigate the carbon footprint, valorization of CO2 into value-added products is of fundamental interest. In this regard, use of CO2 as C1 feedstock for HCHO synthesis would be a more sustainable pathway to pursue than the current petrochemical approach. Yet, this would require development of practical catalytic systems, let alone straightforward access to renewable energy sources. One possibility is to first convert CO2 into syngas, CH3OH, or formate (HCOO−), as these can be used as raw materials for HCHO synthesis. In fact, these processes have caught the attention of many research groups with promising progress reported,4-6 although practical application of CH3OH oxidation and HCOO− reduction to HCHO are yet to be realized. Direct conversion of syngas with a 1 : 1 ratio of carbon monoxide (CO) and hydrogen (H2) to HCHO has been realized in aqueous media.7 It is noteworthy that these processes proceed under harsh conditions, which for large-scale applications would imply significant energy consumption. Thus, development of CO2 conversion processes applicable to ambient conditions is pertinent.
Direct conversion of CO2 to HCHO can be realized using hydrogenation,8, 9 hydroboration,10 hydrosilylation,11 as well as photochemical,12, 13 electrochemical,14, 15 photo-electrochemical,16-18 and enzymatic approaches.19 To minimize overall energy consumption, a reaction powered directly by renewable light and/or electricity would be preferable. Unfortunately, the subsequent conversion of HCHO to CH3OH is strongly exergonic, and it is hard to halt this reaction under these conditions and thus implement it for large-scale production.20 For this reason, most experimental and theoretical chemists refer to HCHO as an important intermediate for CH3OH formation from CO2,21, 22 although with development of efficient trapping strategies, further reduction of HCHO may be avoidable.
In this Minireview, recent progress in the production of HCHO from CO2 is presented and discussed according to catalysis type, i.e., chemical catalysis (hydrogenation using H2 and hydroboration/hydrosilylation), photo/electrocatalysis, and biocatalysis (enzymatic reduction) (Figure 1). From an analysis of advantages and deficits of each methodology, we come up with viewpoints and potential strategies for optimizing CO2-to-HCHO conversion.

Schematic representation of the scope of the Minireview.
2 Chemical Catalysis
2.1 Hydrogenation Using H2
Hydrogenation of CO2 to value-added products is the most atom-efficient route for CO2 transformation. So far, great progress has been made to generate various C1 and C2+ chemicals, including CO,23, 24 methane (CH4),25, 26 CH3OH,27-29 formic acid (HCOOH),30 and hydrocarbons.31-33 Both heterogeneous and homogeneous catalysts were employed for selective conversion of CO2 to HCHO or derivatives thereof. Hydrogenation of CO2 to HCHO [CO2 (g)+H2 (g)→HCHO (g)+H2O (g); Δ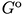 =59.8 kJ mol−1, Δ
=59.8 kJ mol−1, Δ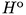 =39.8 kJ mol−1] is a strongly endergonic/endothermic reaction,34 which is thus thermodynamically unfavored under ambient conditions. In addition, catalytic transformation of CO2 with halt at the HCHO level remains challenging, as the resulting aldehyde group is susceptible to further reduction.
=39.8 kJ mol−1] is a strongly endergonic/endothermic reaction,34 which is thus thermodynamically unfavored under ambient conditions. In addition, catalytic transformation of CO2 with halt at the HCHO level remains challenging, as the resulting aldehyde group is susceptible to further reduction.
In 2001, Lee and co-workers reported Pt/Cu/SiO2 as heterogeneous catalyst for the selective hydrogenation of CO2 to HCHO at 150 °C and 6 bar.35 The Pt/Cu-based part, with an optimum atomic Pt/Cu ratio of 0.03 : 1, was found to be crucial for HCHO formation, accompanied by CH3OH as main by-product. Mechanistically, H2 was suggested to be adsorbed on Pt before migrating onto Cu to promote reduction of CO2. Furthermore, the relative rate of HCHO formation increased with the H2/CO2 ratio, showing the strong effect exerted by the surface hydrogen concentration on HCHO selectivity. At an optimum H2/CO2 of 20 : 1, the yield of HCHO reached 10.4 mmol gcat−1 over 2 h reaction where gcat−1 denotes the weight unit of the catalyst. Tanksale and co-workers first demonstrated production of HCHO via hydrogenation of CO2 using Pt/Ni/Al2O3 as catalyst in CH3OH as liquid medium at 25 °C.36 Increasing the H2/CO2 ratio from 2 to 16 led to 50 % increase in the equilibrium HCHO yield, i.e., from 0.06 to 0.09 mmol gcat−1 over 22 h reaction. Despite the limited availability of heterogeneous catalysts for efficient CO2-to-HCHO conversion, a few have been reported in the hydrogenation of CO2 to CH3OH.37 In addition, insight into the underlying mechanism can help to guide the preparation of novel heterogeneous catalysts for hydrogenation of CO2 to HCHO, as surface-bound HCHO derivatives have been demonstrated to be the key intermediate along the CO2-to-CH3OH formation pathway.38, 39
A disadvantage of direct hydrogenation is associated with the need of applying high pressure of H2 to enhance its solubility. In various hydrogenations, liquid-phase alcohols have been used as hydrogen donors, due to higher safety and more convenient management and usage. Recently, Liu and co-workers found that transfer hydrogenation of CO2 into HCHO from aqueous glycerol could be efficiently induced by highly dispersed Ru on layered double hydroxide (LDH) under base-free conditions at low temperature.40 The yield of HCHO could be as high as 3.6 mmol gcat−1 with a turnover number (TON) of 72 over 12 h at 50 °C and 10 bar CO2. The metal–support interaction over Ru/LDH proved to afford high turnover of hydrogen transfer between C3 species (glycerol and lactic acid) and CO2 to generate HCHO. In addition to the direct synthesis of HCHO, conversion of CO2 to dimethoxymethane (DMM), the dimethyl acetal of HCHO, can be realized by heterogeneous catalysts. Recently, Wang, Tanksale, and co-workers synthesized 3 wt % Ru over novel hierarchical zeolite beta to provide an optimized DMM yield of 0.66 mmol gcat−1 after 20 min of reaction.41 Moreover, the activity could be retained even after five recycle steps, demonstrating the reusability of this catalyst.
Homogeneous catalysts in direct hydrogenation of CO2 to HCHO have been less successful than heterogeneous ones. Stephan, Fontaine, and co-workers demonstrated that the frustrated Lewis pairs (FLP) 1-BR2-2-NMe2C6H4 (R=2,4,6-Me3C6H2 or 2,4,5-Me3C6H2) are active for CO2 hydrogenation in the presence of CO2 and H2, generating the corresponding formate, acetal, and methoxide species.42 However, the H2 activation and CO2 reduction steps are stoichiometric and not catalytic in nature.
Several works have studied the mechanistic pathways for CO2 hydrogenation to CH3OH from a theoretical point of view which may provide insights into the reduction of CO2 to the HCHO level, noting once more that HCHO is a key intermediate along the CH3OH production pathway. Klankermayer, Leitner, and co-workers conducted detailed mechanistic studies on the hydrogenation of CO2 to CH3OH using a Ru–triphos (triphos=1,1,1-tris(diphenylphosphinomethyl)ethane) complex.43 The results supported by DFT calculations reveal that a sequential series of hydride transfer and protonolysis steps account for the transformation of CO2 via HCOO−/HCOOH to hydroxymethanolate/HCHO and finally methanolate/CH3OH within the coordination sphere of the complex. Based on DFT calculations, Yang and co-workers predicted a hydride transfer mechanism for the conversion of CO2 and H2 to CH3OH catalyzed by a half-sandwich Co complex, [Cp*Co(bpy-CH3)OH2]2+.44 Three cascade cycles were proposed: hydrogenation of CO2 to HCOOH, hydrogenation of HCOOH to methanediol which decomposes to HCHO and water, and hydrogenation of HCHO to CH3OH. A similar mechanism was proposed by Lei and co-workers for CO2 hydrogenation catalyzed by a Mn pincer complex, Mn(Ph2PCH2SiMe2)2N(CO)2, or a Ru pincer complex, RuH2(Me2PCH2SiMe2)2NH(CO).45, 46 Here, H2 activation is the rate-controlling step in each catalytic cycle under solvent-free conditions. In comparison, the reaction is more favorable in the appropriate solvent (water, toluene, THF) than in the gas phase. These works provide guidelines for how to design new homogeneous catalysts exhibiting higher efficiency for hydrogenation reactions.
Most of the currently available homogeneous catalysts for CO2 hydrogenation have been developed with focus on obtaining DMM as main product instead of HCHO. This is done by performing the hydrogenation reaction in presence of CH3OH, to make the overall process [CO2 (g)+H2 (g)+2 MeOH (l)→MeOCH2OMe (l)+H2O (l); Δ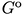 =12.5 kJ mol−1, Δ
=12.5 kJ mol−1, Δ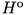 =−73.4 kJ mol−1) exothermic, with an only slightly positive value of the Gibbs energy. The equilibrium towards product becomes even more favored at pressures of 80 bar (Δ
=−73.4 kJ mol−1) exothermic, with an only slightly positive value of the Gibbs energy. The equilibrium towards product becomes even more favored at pressures of 80 bar (Δ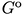 =−0.3 kJ mol−1).34 In 2016, Klankermayer and co-workers reported the synthesis of DMM (Figure 2a) using Ru complex catalysts with triphos ligands (Figure 2b) in combination with acidic co-catalyst Al(OTf)3.47 DMM was obtained with TON up to 214 (over 18 h) at moderate temperature (80 °C) and pressure (60 bar H2/20 bar CO2). In addition, methylformate was isolated in small amounts. Importantly, DMM can be used as a HCHO synthon in syntheses and if desired, DMM may be hydrolyzed to HCHO and CH3OH. The acidic co-catalyst was found to be pivotal in enabling fast acetal formation in the hydrogenation of methylformate to DMM. Formation of the relatively stable acetal prevented over-hydrogenation to CH3OH or CH4. Similarly, other acetals could be synthesized when using other alcohols, such as ethanol and butanol.
=−0.3 kJ mol−1).34 In 2016, Klankermayer and co-workers reported the synthesis of DMM (Figure 2a) using Ru complex catalysts with triphos ligands (Figure 2b) in combination with acidic co-catalyst Al(OTf)3.47 DMM was obtained with TON up to 214 (over 18 h) at moderate temperature (80 °C) and pressure (60 bar H2/20 bar CO2). In addition, methylformate was isolated in small amounts. Importantly, DMM can be used as a HCHO synthon in syntheses and if desired, DMM may be hydrolyzed to HCHO and CH3OH. The acidic co-catalyst was found to be pivotal in enabling fast acetal formation in the hydrogenation of methylformate to DMM. Formation of the relatively stable acetal prevented over-hydrogenation to CH3OH or CH4. Similarly, other acetals could be synthesized when using other alcohols, such as ethanol and butanol.

Overview of homogeneous catalysts used for synthesis of DMM from CO2; Structures are redrawn from the references listed (Tmm=trimethylene methane).
Later, Klankermayer and co-workers developed a tailored non-precious transition-metal catalytic system based on Co salts in combination with selected triphos ligands and acidic co-catalyst (HNTf2) (Figure 2c).48 For example, a TON of 157 (over 22 h) for the formation of DMM, comparable to that of the precious-metal system, was obtained at slightly higher temperature (100 °C), but at the same pressure (60 bar H2/20 bar CO2), and with a modified triphosTol ligand (Tol=toluene). Inspired by this, Trapp and co-workers developed a highly efficient catalytic system using a Ru catalyst with a N-triphosPh (Ph=phenyl) ligand (Figure 2d), raising the TON for DMM to 786 (18 h) with a remarkable selectivity of >90 % at moderate temperature (90 °C) and partial pressure (90 bar H2/20 bar CO2).49 By substituting the apical carbon atom in the backbone of the triphos ligand platform with silicon or phosphorus (Figure 2e–g), they achieved a TON as high as 685 (18 h) for DMM at optimized conditions, being in the range of the benchmark N-triphosPh system.50
Although much progress has already been achieved in CO2 hydrogenation, the transformation of CO2 to the HCHO level still faces great challenges. Numerous mechanistic studies have inferred the intermediacy of HCHO during CH3OH generation in both catalytic homogeneous and heterogeneous processes. Nevertheless, the thermodynamically favored over-reduction of HCHO and its derivatives to the CH3OH level makes it challenging to obtain HCHO via a direct reduction route. To halt the reduction process at this point would require careful manipulation of the reaction conditions, i.e., the chemical environment around the active species, temperature, solvent, reducing agent, etc. Another important aspect is that H2, as one of the main reactants, is predominantly produced using fossil fuels today.51 This results in the so-called CO2-paradoxon in which the CO2 produced for generating H2 exceeds the amount of CO2 consumed in the hydrogenation. Thus, it becomes crucial to sustainably generate carbon-free H2 on a large scale.
2.2 Hydroboration/Hydrosilylation
Looking at the overall energetics of hydroboration or hydrosilylation of CO2, the first important point to note is that hydroborane and hydrosilane reductants feature relatively weak and polarized B−H and Si−H bonds compared with the H−H bond. Second, the CO2 reduction results in formation of strong Si−O and B−O bonds.52 As a result, hydroboration and hydrosilylation of CO2 require significantly milder operating conditions (temperature <100 °C and CO2 pressure <3 bar) than use of H2 while enabling the synthesis of the complete series of C1 products (CO, HCHO, CH3OH, CH4). The exact distribution of these products depends on the nature of the catalyst introduced for regenerating the hydroborane/hydrosilane reductants.
Taking the commonly used metal-based catalysts as an example, CO2 hydroboration usually involves three sequential catalytic cycles (Figure 3). In Cycle I, CO2 is reduced to the HCOOH level, in the form of R2B−O(O)CH. This involves initial CO2 insertion into the metal hydride (M−H), followed by transmetalation of M−OOCH with hydroborane (HBR2) to regenerate M−H and release R2B−O(O)CH. In Cycle II, further reduction leads to the HCHO level. Specifically, M−H reacts with R2B−O(O)CH, resulting in M−OCH2OBR2, which is decomposed to M−OBR2 and HCHO. Meanwhile, M−OCH2OBR2 can transmetalate with HBR2 to generate bis(boryl)acetal (R2BOCH2OBR2) and M−H feeding once again into Cycle I. Cycle III is the CH3OH level with formation of CH3OBR2, starting with insertion of HCHO into M−H to generate M−OCH3 that reacts with HBR2.

Proposed pathway for CO2 hydroboration using metal-based catalysts in which products at the HCOOH (boryl formate), HCHO [bis(boryl)acetal], and CH3OH (methoxy borane) oxidation levels are obtainable. Note that methoxy borane formation results in production of an equivalent of bis(boryl)oxide; Figure is redrawn from ref. 58; Copyright 2018, American Chemical Society.
Hydrosilylation using metal-based catalysts follows an equivalent pathway to that of hydroboration, although a fourth cycle now extends the product portfolio to CH4. Usually, the main products of CO2 hydroboration or hydrosilylation are formoxy (Cycle I; single reduction) or methoxy (Cycle III; triple reduction). Selective reduction of CO2 to the HCHO level (Cycle II; double reduction) is more challenging. Only by carefully manipulating reaction conditions (temperature, CO2 pressure, concentration, etc.) is it possible to halt the cascade of reduction processes at the HCHO level.
Sabo-Etienne and Bontemps pioneered the catalytic reduction of CO2 using pinacolborane (HBpin) as reductant and RuH2(η-H2)2(PCy3)2 (Cy=cyclohexyl) as catalyst precursor to obtain HCHO in the form of bis(boryl)acetal, PinBOCH2OBPin, with a low yield of 5 % and low TON of 0.5 over 30 min of reaction at room temperature (Figure 4a1).53 CH3OH was further used to capture HCHO from the reaction by forming CH3OCH2OH.54 Later, they disclosed formation of free HCHO (yield=22 %, TON=2.2), along with bis(boryl)acetal (yield=6 %, TON=0.6) over 30 min of reaction, using the analogous complex [RuH2(H2)2(PCyp3)2] (Cyp=cyclopentyl) as catalyst precursor (Figure 4a2).55 The better performance of the cyclopentyl-based catalyst was attributed to differences in cycloalkyl ring conformations and solubilities. Selective trapping of HCHO was realized by in situ condensation with 2,6-bis(diisopropyl)aniline to afford the corresponding imine. Both the yield of the corresponding imine and TON (74 % and 10.6, respectively) over 1 h of reaction were greatly improved by the presence of the trapping agent. In addition, subsequent hydrolysis of the imine with CH3OH regenerated aniline and yielded a formalin solution.

Overview of catalysts employed for CO2 hydroboration and hydrosilylation together with the specific hydroborane or hydrosilane reagents used in each case; Structures are redrawn from the references listed.
In addition to the Ru-based catalysts, they realized Fe-catalyzed reduction of CO2 into bis(boryl)acetal using Fe(H)2(dmpe)2 (dmpe=Me2PCH2CH2PMe2) (Figure 4a3).56 Three different hydroboranes were considered, i.e., HBpin, catecholborane (HBCat), and 9-borabicyclo[3.3.1]-nonane (9-BBN). With HBCat, only methoxyborane was obtained while with HBpin and 9-BBN, the corresponding bis(boryl)acetal was favored over methoxyborane. Optimization of the synthesis using 9-BBN as reduction agent and tetrahydrofuran as solvent provided an excellent yield of 85 % with a TON of 85 for bis(boryl)acetal over 47 min. Inspired by this work, Turculet and co-workers reported a new bis(indolylphosphino)silyl (PSiP) ligand (Figure 4a4), the Ni hydride complex of which exhibited an unprecedented yield of 97 % for hydroboration of CO2 to the targeted bis(boryl)acetal, with a TON of 487 after 4 h of reaction under 1 atm CO2 at room temperature.57 Hazari and co-workers performed a systematic study on how to control the reduction level of CO2 hydroboration using the Ni−PSiP complex (Figure 4a4) and (tBuPCP)−NiH [tBuPCP=2,6-C6H3(CH2PtBu2)2] (Figure 4a5).58 They demonstrated that reduction of CO2 past the HCOOH level could be accomplished by decreasing the CO2 concentration, increasing the hydroborane concentration, or adding a Lewis acid cocatalyst. With this, borylformate insertion into M−H (Cycle II) became favored over the corresponding CO2 insertion (Cycle I). In addition, low catalyst loading resulted in precipitation of bis(boryl)acetal, providing a driving force for its formation. Xu and co-workers found that a molecular ZnII dihydride complex served as active catalyst for CO2 hydroboration (Figure 4a6), selectively generating bis(boryl)acetal with a yield of 75 % and a TON of 15 after 4 h reaction using 9-BBN as hydroboration reagent.59
Leitner and co-workers reported the Mn pincer complex, Mn(Ph2PCH2SiMe2)2NH(CO)2Br, that enables reduction of CO2 in the presence of hydroboranes.60 Lei, Cao, and co-workers investigated the reaction mechanism of CO2 hydroboration catalyzed by the Mn pincer complex.61 The calculated results showed that the carbonyl association mechanism is more favorable, with a low energetic span of 113 kJ mol−1, than the carbonyl dissociation mechanism. The intrinsic difference between these two pathways is whether the second CO ligand coordinates with the metal center or not. The carbonyl dissociation mechanism mainly includes three catalytic cycles: hydroboration of CO2 to yield the intermediate product HCOOBpin, hydroboration of HCOOBpin to yield HCHO and O(Bpin)2, and hydroboration of HCHO to yield methyl boronate (CH3OBpin). Regarding the same Mn pincer complex, Li, Schaefer, and co-workers reported a novel ionic mechanism for CO2 hydroboration in the presence of sodium tert-butoxide (NaOtBu).62 The ionic mechanism also contains three tandem stages. Differently, the H−Mn−N−Bpin pincer species generated by the reaction of the Mn pincer complex and HBpin in the traditional mechanism is not viable under basic conditions due to abstraction of the Bpin group by NaOtBu, while the generated H−Mn−N−Na pincer species leads to a more favorable ionic mechanism. In comparison, the energetic span of the ionic mechanism is calculated to be 94 kJ mol−1 lower than that of the traditional mechanism. Altogether, these experimental and computational works suggest that the Mn pincer complex is a promising catalyst for CO2 hydroboration to the HCHO level.
With regard to hydrosilylation, Piers, Maron, Eisenstein, and co-workers reported selective reduction of CO2 to the HCHO level using the [Cp2Sc][HB(C6F5)3] ion pair (Figure 4b1).63 Berke and co-workers demonstrated a Re−H/B(C6F5)3 Lewis pair catalyst (Figure 4b2), which was able to reduce CO2 to (Et3SiO)2CH2 within 4 h with a yield of 35 % and TON of 35.64 Later, Piers and co-workers designed a novel organoscandium-based hydrosilylation catalyst (Figure 4b3) which, when combined with B(C6F5)3, was capable of effectively hydrosilylating CO2 almost exclusively to the bis(silyl)acetal, (Et3SiO)2CH2, with an isolated yield of 94 % and TON of ∼1000 for 96 h in the presence of Et3SiH.65 Oestreich and co-workers reported Ru-catalyzed hydrosilylation of CO2 yielding silylated HCHO with a high yield of 99 % and TON of 25 over 4 h using Et3SiH as hydrosilane at relatively low temperature (80 °C) (Figure 4b4).66 After additional 7 days at 150 °C, 77 % of the formed silylated HCHO were converted to silylated CH3OH, indicating that the oxidation level of the product (silylated HCHO or CH3OH) could be readily controlled by temperature.
López-Serrano, Rodríguez, and co-workers found that a bis(phosphino)boryl Ni−H complex (Figure 4b5), in combination with B(C6F5)3, selectively hydrosilylated CO2 to the bis(silyl)acetal, (Et3SiO)2CH2, with a yield of 60 % and TON of 1200 (21.5 h reaction time) using Et3SiH as hydrosilylation agent.67 Mechanistic studies demonstrated that the metal complex is instrumental in the reduction steps during catalysis via FLP-like bond activation steps involving silylium transfer from [R3Si−H⋅⋅⋅B(C6F5)3]. This is followed by hydride transfer from [HB(C6F5)3]− at contact ion pairs such as [(PBP)Ni][HB(C6F5)3], which is the species responsible for CO2 activation. In addition, the metal complex effectively sequesters B(C6F5)3, thus limiting the amount of [R3Si−H⋅⋅⋅B(C6F5)3] available in the reaction medium, and preventing the latter species from catalyzing the over-reduction of bis(silyl)acetal (Figure 5).68 Parkins and co-workers reported that terminal Zn and Mg hydride compounds (Figure 4b6) were capable of hydrosilylation of CO2 by hydrosilanes at room temperature, when reacting with B(C6F5)3 to afford the ion pairs.69 In the presence of Ph3SiH, the Zn complex could reduce CO2 to (Ph3SiO)2CH2 with a TON of 12 over 12 h, while the Mg complex selectively afforded (Ph3SiO)2CH2 with a TON of 83 over 70 h. In general, hydrosilylating CO2 to the bis(silyl)acetal level was favored using a sterically hindered hydrosilane, e.g., Ph3SiH, while PhSiH3 would form CH4. A N,P-heterocyclic germylene/B(C6F5)3 Lewis adduct (Figure 4b7), reported by Baceiredo, Kato, and co-workers, was able to hydrosilylate CO2 exclusively to (Et3SiO)2CH2 with a TON of 19.9 (25 h reaction time) at 60 °C.70 In 2019, Parkin and co-workers demonstrated that (Ph3SiO)2CH2 could be generated using a Mg complex (Figure 4b8) from CO2 hydrosilylation with an isolated yield of 95 % and TON of 88.6. In addition, HCHO was released from (Ph3SiO)2CH2 upon treatment with CsF at room temperature.71

Werlé, Leitner, and co-workers demonstrated the possibility of directing the hydrosilylation of CO2 to either of the HCOOH, HCHO, or CH3OH levels by adjusting temperature, solvent, and pressure when using a Co triazine pincer complex as catalyst (Figure 4b9).72 In general, higher temperature, lower CO2 pressure, and higher catalyst concentration would favor HCHO formation. Optimally, a yield of 50 % and TON of 56 for HCHO derivatives could be obtained under solvent-free conditions (2.5 mmol silane, 0.2 mol % Co, 0.8 mol % potassium tert-butoxide, 80 °C, 1 bar, 4 h reaction time). Computational results demonstrated that the kinetic energy barrier for the catalytic formation of silyl formate (cycle I, 101 kJ mol−1) is smaller than that of the formation of bis(silyl)acetal (cycle II, 121 kJ mol−1) and that of the formation of methoxysilane (cycle III, 125 kJ mol−1).73 It rationalizes the need of using higher temperature for reducing CO2 to the HCHO and CH3OH derivatives. In addition, the similar energy barrier of Cycles II and III indicates that it is difficult to avoid the over-reduction to CH3OH derivatives. Despite this, the calculated high kinetic hindrance associated with the rearrangement of bis(silyl)acetal to HCHO and 1,3-diphenylsiloxane is identified as crucial to selectively halt the reaction at the HCHO level.
Aldridge and co-workers found that a 3-coordination cationic gallane complex (Figure 4b10), partnered with a hydroborate anion, catalyzed CO2 reduction selectively to the HCHO level with a TON of 295.8 over 58 h in the presence of Et3SiH at 60 °C.74 Recently, Maron, Xu, and co-workers observed selective generation of bis(silyl)acetal without over-reduction in the catalytic system of homoleptic La aryloxide and B(C6F5)3 (Figure 4b11).75 An exceedingly high yield of 99 % was reached, corresponding to a TON of 670 (13 h reaction time), with the bis(silyl)acetal (Ph3SiO)2CH2 formed upon using the sterically hindered Ph3SiH. Thus, the reaction outcome depended highly on the nature of the silane reductant.
While use of metal complexes as catalysts has dominated the hydroboration/hydrosilylation field, organocatalysts have begun to emerge as more cost-effective and ecologically friendly alternatives for CO2 reduction. Zhang, Ying, and co-workers reported that N-heterocyclic carbenes promoted the formation of bis(silyl)acetal from hydrosilylation of CO2 using diphenylsilane as reducing agent.76 Unlike the mechanistic pathways shown in Figure 3, the CO2 reduction was suggested to be initiated by nucleophilic attack of the carbene on CO2, forming an imidazolium carboxylate. The carboxy moiety would then attack the electropositive silane center and promote hydride transfer to afford the key intermediate, formoxysilane.
Later, Maron, Fontaine, and co-workers found that the ambiphilic organocatalyst 1-Bcat-2-PPh2C6H4 (cat=catechol) can efficiently reduce CO2 to CH3OBR2 in presence of hydroboranes, with the HCHO derivative acting as key intermediate.77 Mechanistic investigation revealed that hydroboration occurred through simultaneous Lewis base activation of the hydroborane and Lewis acid activation of CO2.78 In addition, activation of the HBcat moiety by the phosphorus center, while the substrate was fixed and activated by the Lewis acidic boron center, reduced the entropic cost of the catalyzed steps (reductions of HCOOBcat and HCHO) (Figure 6).

Proposed mechanistic pathway (including important transition states) for hydroboration of CO2 by 1-Bcat-2-PPh2C6H4. Reproduced from ref. 78; Copyright 2018, American Chemical Society.
Cantat and co-workers revealed that two nitrogen bases, i.e., 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (Me-TBD), were active organocatalysts for hydroboration of CO2, although they followed two distinct mechanisms.79, 80 Notably, the N−H bond of TBD is reactive towards dehydrocoupling with hydroborane and affords a frustrated Lewis pair that can activate CO2. This mechanism is similar to that of 1-Bcat-2-PPh2C6H4.77 In contrast, Me-TBD lacks the N−H moiety and promotes CO2 reduction through hydroborane activation.
Compared with the hydrogenation of CO2 using H2, hydroboration/hydrosilylation can be conducted under much milder conditions with respect to both temperature (<100 °C) and pressure (<3 bar). This facilitates sequential synthesis of value-added compounds from HCHO, let alone mechanistic investigations. An interesting opportunity to explore would be using the HCHO intermediates as methylene transfer agents to generate organic compounds such as amines, as the bis(boryl)acetals thus produced would have value in synthetic chemistry in their own right. On the more problematic side is the high cost of hydroborane and hydrosilane reagents, along with production of stoichiometric waste products (after treatment of the carboxylated borane product with acid to generate HCOOH, HCHO, or CH3OH). This sums up to large economic and ecological deficiencies of these processes.
3 Photo/Electrocatalysis
3.1 Photochemical Reduction
Photochemical reduction of CO2 to useful chemicals presents an appealing way of converting sustainable solar energy to chemical energy while helping to mitigate CO2 accumulation. Figure 7a illustrates how illumination of a photocatalyst with incident light excites electrons from the valence band (VB) to the conduction band (CB), leaving an equal number of holes in the VB. While the photogenerated electrons can drive the reduction of absorbed CO2, the holes oxidize water to molecular O2. Both homogeneous molecular and heterogeneous semiconductor catalysts have been widely investigated for photocatalytic CO2 reduction. Product-wise, the focus has been primarily on CO, ethylene, and ethanol.

a) Schematic representation of photochemical (PC) reduction. b) Quantity of HCHO produced during CO2 reduction reaction over CeO2-based photocatalysts determined by gas chromatography; Figure reproduced from ref. 91; Copyright 2020, Elsevier B.V. c) Schematic representation of electrochemical (EC) reduction. d) FE of products generated in eCO2RR using various electrodes in CH3OH electrolyte; Figure reproduced from ref. 102; Copyright 2014, John Wiley and Sons. e) Schematic representation of photoelectrochemical (PEC) reduction of CO2. f) Normalized FE of HCHO plotted against potential in the 040-BVO|NaCl|Cu system; Figure reproduced from ref. 107; Copyright 2017, American Chemical Society.
In 1979, HCHO was first detected as a photocatalytic product of CO2 using photosensitive semiconductor powders (TiO2 and SiC) suspended in water as catalysts.81 The suspensions were illuminated for 7 h under a 500 W Xe lamp with a cut-off filter (<500 nm). Yields were determined to be 0.11 mmol gcat−1 for HCHO and 0.023 mmol gcat−1 for CH3OH using the TiO2 catalyst, and 0.1 mmol gcat−1 for HCHO and 0.54 mmol gcat−1 for CH3OH with SiC. Since then, TiO2 was regarded as one of the best photocatalysts for CO2 photoreduction because of its high activity and long lifetime. However, high electron–hole recombination rate and limited absorption in the visible region are severe drawbacks, although these can be alleviated by modifying TiO2 using noble metals through interband electron transition (Figure 7a, right). Thus, a series of Au/TiO2-based catalysts with low Au loading (0.1–0.5 wt %) were tested for CO2 photoreduction in the liquid phase over 24 h of continuous irradiation (77 W Hg lamp with a range of emission wavelengths from 254 to 364 nm) at 7 bar pressure at 80 °C.82 Major products detected in the liquid phase were CH3OH, HCOOH, and HCHO, formed in a distribution depending on the exact catalyst formulation used. With 0.1 wt % Au/TiO2, maximum productivity of HCHO of 0.19 mmol g−1 h−1 was obtained. In another work, Au-NP-decorated TiO2 nanotube arrays (Au−PMTiNTs) were used as photocatalyst to selectively convert CO2 to CO (0.32 mmol g−1 h−1) and HCHO (0.42 mmol g−1 h−1) under a 50 W white LED (Pmax at 520 nm) for 2 h, followed by irradiating the reactor with simulated sunlight for 2 h.83 In other studies with TiO2 as catalyst, typically either a low selectivity of HCHO15 or further conversion of HCHO to CH3OH86, 87 was reported.
In 2013, the design of a bifunctionalized TiO2 film containing dye-sensitized and catalysis zones was reported for the visible light photocatalytic reduction of CO2 under a 300 W Xe lamp (λmax=500 nm).86 In the first two hours reaction time, the main products were HCOOH and HCHO, but thereafter the yield of CH3OH increased sharply because of the preceding buildup of HCHO. This strongly suggests that the stepwise pathway of CH3OH production proceeding via reduction of HCOOH and HCHO is more favorable than a one-step conversion of CO2 to CH3OH. Ananthakrishnan and co-workers introduced benzimidazole-containing Ru metal complexes with two different functional groups (−COOH and −NO2) for TiO2 sensitization.88 The hybrid catalyst with the -COOH group showed excellent photocatalytic activity towards CO2 reduction, with a HCHO production rate of 1.1 mmol g−1 h−1 under irradiation of a 250 W tungsten lamp (>420 nm) for 5 h.
In addition to TiO2, CeO2 is also a good candidate as photocatalyst for CO2 reduction, which can activate CO2 by adsorbing and deforming its linear molecular structure.89, 90 Kang and co-workers reported a NiO/CeO2/rGO (reduced graphene oxide) hybrid composite photocatalyst to produce HCHO selectively (0.42 mmol g−1 h−1) under illumination of a 300 W Xe lamp with a simulated sunlight wavelength for 5 h, around four times faster than if pristine CeO2 was used (Figure 7b).91 The high CO2 reduction ability was ascribed to formation of a p–n junction at the NiO−CeO2 interface, as this would suppress charge carrier recombination. Furthermore, introduction of rGO allowed accumulation of electrons at the hybrid composite photocatalyst surface with subsequent transfer to the activated CO2. A carbon-doped NaTaO3 perovskite annealed at 650 °C presented a HCHO production (0.02 mmol g-1 h-1) under irradiation by a pen ray lamp (UV, 254 nm) for 2 h.84
In general, homogeneous molecular catalysts pertaining to Ru, Os, Ir, and Co complexes are known to provide good quantum yields for CO2 reductions.92, 93 In the specific case of HCHO as product, Chatterjee and co-workers reported a suitable system consisting of K[Ru(H-EDTA)Cl]⋅2 H2O as homogeneous catalyst and particulate Pt−CdS−RuO as photon absorber at 505 nm.94 Here, the photocatalytic reduction at a constant (dissolved) CO2 concentration (3.38×10−2 M) in water, irradiated at 505 nm using a 250 W Xe lamp, produced 0.10 M of HCHO along with 0.22 M of HCOOH during 6 h photolysis at rates of 3.05×10−2 and 2.0×10−2 M h−1, respectively.
Currently, state-of-the-art photocatalysts consist of semiconductors with appropriate band gap or homogeneous catalysts such as the costly Ru complexes. The first challenge of CO2 photoreduction is the low solubility of CO2 in aqueous solution. Although low solubility can be mitigated by increasing temperature or pressure as reported in some works, this would soon show its costs on the energy account. Second, the selectivity towards HCHO is modest. Third, the quantum yield of the photocatalytic process is comparably low. This low utilization rate of solar energy calls for development of more active and efficient photocatalysts before large-scale application can come true.
3.2 Electrochemical Reduction
Electrochemistry has emerged as a promising platform for CO2 reduction with electrons from the electrode serving as reductants (Figure 7c). Such reductions can usually be conducted under ambient conditions without necessity of adding chemical reductants. The electrochemical CO2 reduction reaction (eCO2RR) has been capable of generating CO, HCOO−, hydrocarbons, and oxygenates in high yields and selectivity using various kinds of metal-based catalysts, single-atom catalysts, and molecular catalysts.95-99
In contrast, reports on HCHO have been relatively scarce because of the difficulty in preventing its further reduction once formed. Nevertheless, HCHO is often identified as minor product (<10 %) in eCO2RR. In 2014, Dyer and co-workers reported a pterin (natural cofactor for a wide range of enzymes) electrocatalyst, 6,7-dimethyl-4-hydroxy-2-mercaptopteridine (PTE), which catalyzed the reduction of CO2 on a reticulated vitreous carbon electrode to HCOOH, HCHO, and CH3OH.100 It was suggested that PTE acted as a multifunctional catalyst for converting CO2 to HCOOH, followed by further reduction to HCHO or other products. Later, Proust and co-workers used (TOA)6[α-SiW11O39Co(_)] (TOA=tetraoctyl ammonium; _=vacant position in the coordination sphere of Co) as homogeneous electrocatalyst and achieved a maximum faradaic efficiency (FE) for HCHO formation of 40 % at −1.5 V vs. saturated calomel electrode (SCE) in CH2Cl2.101 Unfortunately, CH2Cl2 as solvent greatly limits practical applications due to its high volatility and potential degradation (dehalogenation) during electrolysis.
Nakata, Einaga, and co-workers discovered boron-doped diamond (BDD) as an efficient electrocatalyst in CO2-to-HCHO conversion under ambient conditions with a FE of 74 % in CH3OH electrolyte and 62 % in 0.1 M NaCl aqueous electrolyte at −1.5 V vs. Ag/Ag+ (Figure 7d).102 Notably, HCOOH came out with FE <15 % and hydrogen evolution was, likewise, of small concern. Compared with glassy carbon electrodes consisting of sp2-bonded carbons, BDD as electrode material showed much better selectivity for HCHO, inferring that the sp3-bonded carbons on BDD might be the key factor. Yet, current densities were exceedingly low (in the μA cm−2 range), calling for further performance improvements by, e.g., increasing the number of active sites and tuning the electronic structure of BDD.
Recently, Wang, Liang, and co-workers reported a Co β-tetraaminophthalocyanine molecular catalyst supported on carbon nanotubes which converted CO2 and NO3− to methylamine in aqueous media under ambient conditions.103 Methylamine was produced at a maximum FE of 13 % with a partial current density of 3.4 mA cm−2 at −0.92 V vs. reversible hydrogen electrode (RHE). At first, electrochemical reductions of CO2 and NO3− proceed independently with formation of HCHO and NH2OH, but soon adsorbed HCHO is to undergo nucleophilic attack by NH2OH. This yields formaldoxime, which is reduced to N-methylhydroxylamine and further to methylamine. Later, the authors demonstrated that addition of different nitrogen sources, i.e., amine, hydrazine, hydroxylamines, and nitro compounds, could be feasible for electrochemical reductive N-methylation.104 This in situ trapping approach can broaden the adoption area of suitable electrocatalysts capable of producing HCHO as intermediate, in spite of the challenges persisting in halting the process at the HCHO level under reductive electrochemical conditions.
Although electrochemical reductions present one of the most sustainable approaches for producing HCHO from CO2, the number of electrocatalysts capable of realizing this transformation is quite limited. Moreover, currently reported catalysts are met with the serious drawbacks of having both low activity and selectivity. More investigations are therefore needed to improve the overall performance by increasing the number of active sites and tuning the electronic structure.
3.3 Photoelectrochemical Reduction
Photoelectrochemical reduction aims to combine the best of the photochemistry and electrochemistry worlds. While the photoelectrode generates electron–hole pairs upon light irradiation, the applied bias potential drives the electrons to the cathode for CO2 reduction (Figure 7e). As a result, the recombination probability of photogenerated electron–hole pairs diminishes while the electron–hole transfer to their respective acceptors accelerates, which maximizes the overall yield. In addition, the photovoltage gained at the semiconductor/solution interface can be utilized to generate an underpotential, reducing the required bias potential to activate CO2.
In 1978, Halmann performed the first study of this kind on CO2 reduction using p-type Ga phosphide as photocathode.105 After 18 h irradiation under a high-pressure mercury arc with no filters and application of a cathodic bias of −1.0 V vs. SCE, HCOOH was produced as main product (1.2×10−2 M), accompanied by a minor amount of HCHO (3.2×10−4 M) and CH3OH (1.1×10−4 M). Similarly, low HCHO selectivity was the result of many subsequent investigations.16-18, 106 Then, in 2018, Kang and co-workers reported an efficient system combining a (040)-facet engineered BiVO4 plate photoanode (040-BVO) and a Cu cathode in NaCl electrolyte (Figure 7f).107 The maximum FE achieved for HCHO was 85.1 % at a bias potential of −0.9 V vs. RHE under irradiation by a 300 W Xe arc lamp with simulated sunlight. Recently, they designed a Ca–Fe doped TiO2 photoanode and a Cu cathode modified using, as minimum, rGO.108 With this system, HCHO was the major product over Cu/rGO with a FE of 25 %, while acetaldehyde became the major product over a Cu/rGO/polyvinylpyrrolidone/Nafion cathode with a FE of 39 % at −0.68 V vs. RHE bias potential under solar simulated radiation for 8 h.
Photoelectrochemical reduction is one of the most interesting technologies proposed for CO2 conversion. Promising results can be expected with more efforts directed towards the development of photoelectrodes and optimization of reactor configurations.
4 Biocatalysis
Enzymatic reduction of CO2 displays several notable features, owing to high selectivity, mild reaction conditions, and environmental friendliness. Normally, it is a multi-enzymatic, multistep reaction using three dehydrogenases, i.e., HCOO− dehydrogenase (FDH), formaldehyde dehydrogenase (FADH), and alcohol dehydrogenase (ADH). The role of FDH is to convert CO2 to HCOOH which is then reduced to HCHO by FADH. In the last step, HCHO is converted to CH3OH by ADH. All three enzymes use the cofactor nicotinamide adenine dinucleotide as electron donor.
On the basis of this approach, Liu and co-workers demonstrated enzymatic reduction of CO2 to HCHO, employing FDH and FADH as sequential catalysts.109 By carefully studying the reaction kinetics, they found that higher CO2 pressure and larger ratio of FDH to FADH accelerated the reduction process. Nabavi, Zadeh, and co-workers co-immobilized FDH and FADH in mercaptopropyl-modified siliceous mesostructured cellular foams, exhibiting four times increased activity for HCHO production (0.024 mmol genzyme−1 min−1, estimated from the pertinent figure) compared with that in solution (0.007 mol genzyme−1 min−1).110 The co-immobilization method was proposed to place the two enzymes close to each other, thereby resulting in better CO2-to-HCHO conversion.
For biocatalysis, the low activity of FDH and FADH presents a major challenge in CO2 reduction. To enhance the efficiency of the multienzyme cascade reaction, manipulation of the structures and compositions of enzymes to achieve more active FDH and FADH with high turnover rate and modulation of the relative spatial distance between FDH and FADH are promising strategies. Unfortunately, the reversibility of the reaction impedes further improvement of catalytic performance, as reduction of CO2 is easily overwhelmed by the reverse reaction during HCHO accumulation. Thus, immediate removal of generated HCHO from the reaction system by adding trapping agents would be crucial for continuous production of HCHO from CO2.
5 Conclusions and Future Perspectives
5.1 Issues That Need to Be Resolved
HCHO is a vital platform chemical used for producing value-added products such as resins, polymers, and adhesives by the chemical industry. Compared with traditional strategies to produce HCHO, based on partial oxidation/dehydrogenation of CH3OH, catalytic reduction of CO2 presents a sustainable, yet highly challenging pathway. In this sense, selective generation of HCHO utilizing CO2 under mild reaction conditions remains an important goal in chemistry.
The last few decades have witnessed intense research in the CO2-to-HCHO conversion using hydrogenation, hydroboration/hydrosilylation as well as photochemical, electrochemical, photoelectrochemical, and enzymatic reductions. However, despite these advancements, significant issues remain to be addressed carefully before industrial applications can be even considered. In general, hydrogenation of CO2 includes three cascade catalytic cycles: CO2 hydrogenation to the HCOOH level, HCOOH hydrogenation to the HCHO level, and HCHO hydrogenation to CH3OH. The key issue is how to halt the reduction of CO2 at the HCHO level, i.e., achieve quantitative conversion of HCOOH while suppressing further reduction of the produced HCHO. Unfortunately, the low energy barrier of the latter reduction process makes this task exceedingly difficult, although alteration of the energy span for the HCHO and CH3OH levels can be realized by modulating the reaction conditions, i.e., temperature, solvent, CO2 pressure, concentrations, etc.
In the cases of electrochemical and photochemical reduction, the situation often turns out to be more convoluted due to the possibility of having proton-coupled electron-transfer processes. Along with this come CO2 mass transport limitations in aqueous solution because of the relatively low solubility of CO2, resulting in low conversion rates and low selectivity. Encouragingly, utilization of gas diffusion electrodes and flow-cell reactors has, by and large, mitigated the gap between laboratory discovery and industrial needs in this respect.
A point, often considered trivial but important, is the procedure used for HCHO quantification. HCHO is generated in small quantities with low selectivity, and under aqueous conditions, hydrated or oligomeric forms of HCHO exist alongside free HCHO, making its quantification challenging. Various techniques have been applied, including high-performance liquid chromatography (HPLC),111 gas chromatography,108 fluorimetry,112 nuclear magnetic resonance,113 and UV/Visible spectroscopy.7 Notably, pretreatments of HCHO are always necessary in order to form more easily detectable derivatives using reagents such as 2,4-dinitrophenylhydrazine,106 3-methyl-2-benzothiazolinone hydrazine,40 acetoacetanilide,7 2,4-pentanedione,101 and sodium bisulfite.104, 114 HPLC based on hydrazine derivatization is the most frequently used method, as the detection limit is exceedingly low (3 nM). However, the hydrazine agents may also react with NO, NO2 in air, and other aldehydes, decreasing the accuracy and selectivity for HCHO quantification.115
The Hantzsch reaction, based on derivatization of formaldehyde with β-diketone (acetoacetanilide, 2,4-pentanedione), comes with good sensitivity and selectivity for HCHO, but with a much lower reaction rate.116 The chromotropic acid method requires heating of the sample under strongly acidic conditions, which is undesirable in many applications.117 Recently, sodium bisulfite has proved to be an efficient agent for HCHO quantification and H13CHO identification using NMR.114 The drawback of this method is the reactivity of sodium bisulfite itself towards CO2. Consequently, the design of even more specific agents would be of great interest to realize a fast, low-cost, selective, and highly sensitive analysis of HCHO.
5.2 Strategies for CO2-to-HCHO Conversion
Given the rarity of chemical platforms that can efficiently and selectively reduce CO2 to the HCHO level, development of new catalysts would be of considerable importance from both an energy cost-efficiency and environmental perspective. Considering that numerous catalytic systems are known already to efficiently reduce CO2 to HCOOH or CH3OH, they would be worth looking at in a first approach, once appropriately modified and once reaction conditions have been carefully adjusted, i.e., temperature, CO2 pressure, reductant type, concentration, etc.
Among the reported catalysts, organocatalysts present a promising research field for achieving high selectivity towards HCHO. In general, these catalysts together with hydroboranes or hydrosilanes provide diverse mechanistic pathways for reduction of CO2, which offers more chances of halting the cascade of reduction processes at the HCHO level. Although currently available organocatalysts are yet to arrive at a stage where this can be done efficiently, a more elaborate catalyst design, integrating both the Lewis acid and base into a single molecule, would enable direct manipulation of the binding strength of specific intermediates, let alone reduce the entropic cost of the catalyzed step.
In situ trapping of HCHO provides an interesting approach for diverting unwanted reactions and halting CO2RR at the HCHO level. In hydroborations and hydrosilylations, trapping agents have already enabled the synthesis of complex molecules through formation of new C−C, C−N, C−O, and C−P bonds. Much of this work could easily be extended to electro- or photochemical reductions with the notion that judicious choice of the trapping agent with respect to the catalyst system would be vital to avoid further reduction. As mentioned, the biocatalysis field could also benefit greatly from introducing such approaches.
In general, mechanistic investigations are indispensable during the optimization of catalyst structures. In fact, without a fundamental understanding of the various mechanistic pathways and their energetics as well as identification of key intermediates and/or transition states, optimization of reaction conditions would have to rely on a trial-and-error approach to a large extent. In the quest towards mechanistic understanding, use of both theoretical studies and in situ/operando tools becomes indispensable. To date, research on theoretical models for HCHO formation is too rare to provide precise guidance for experimental operation and should be further developed.
Application of in situ/operando characterization techniques in catalytic CO2-to-HCHO conversion is highly desirable, as any short-lived intermediates in the stepwise reaction kinetics will be left undetectable by ex situ characterizations. Recently, various techniques of this kind based on X-ray diffraction/scattering, X-ray photoelectron spectroscopy, X-ray absorption spectroscopy, Raman spectroscopy, infrared spectroscopy, and transmission electron microscopy have been used in CO2RR to resolve many unsettled issues.118, 119 In future work, much more attention should be given to the HCHO formation step using these important techniques to provide clearer guidance for catalyst design based on a thorough mechanistic understanding.
Acknowledgements
This research was supported by the Novo Nordisk Foundation CO2 Research Center (grant no. NNF21SA0072700). Xin-Ming Hu acknowledges the support of the National Natural Science Foundation of China (grant no. 22109089). Hong-Qing Liang is grateful for a Humboldt Research Fellowship (Alexander von Humboldt Foundation). Simin Li thanks the Natural Science Foundation of China (grant no. 52204327).
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Siqi Zhao obtained her BSc (2015) from the Beijing University of Chemical Technology and her MSc (2018) from Soochow University in China. She is currently a PhD candidate at Aarhus University under supervision of Prof. Kim Daasbjerg. Her current work focuses on electrochemical CO2 reduction and flow cell applications.

Biographical Information
Hong-Qing Liang received his BSc (2011) and PhD degrees (2016) from Zhejiang University. He then carried out postdoctoral research at University of Texas at San Antonio (2016–2018), Aarhus University (2018–2019), and University of Padova (2020–2021). Currently, he works as an Alexander von Humboldt fellow at Leibniz Institute for Catalysis in the group of Prof. Matthias Beller and Prof. Robert Francke. His research focuses on novel catalytic systems for electrochemical CO2 reduction.

Biographical Information
Xin-Ming Hu received his BSc and MSc degrees in China from Nankai University in 2008 and Graduate University of Chinese Academy of Sciences in 2011, respectively. He obtained the PhD degree in 2014 from University of Copenhagen, and afterwards worked as a Postdoctoral Fellow and Assistant Professor at Aarhus University, Denmark. In 2020, he moved back to China and joined Shandong University as Professor. His research focuses on the development of materials and technologies for CO2 capture and utilization.

Biographical Information
Simin Li received her PhD degree (2021) at the Interdisciplinary Nanoscience Center (iNANO)/Department of Chemistry at Aarhus University, under supervision of Prof. Kim Daasbjerg. She is currently an Assistant Professor in the School of Metallurgy and Environment at Central South University in China, in the group of Prof. Jie Li and Prof. Yanqing Lai. Her current research focuses on the development of solid-state batteries and advanced functional materials for lithium/sodium ion batteries

Biographical Information
Kim Daasbjerg obtained his PhD in 1993 working on organic electrochemistry under supervision of Prof. Henning Lund at Aarhus University. He achieved a Doctor of Science degree in 2006 and was promoted to Professor at Aarhus University in 2010. In addition to electrochemistry, his expertise includes surface modification tools as applied to the study of polymer brushes, responsive polymers, coatings, and smart hybrid materials. More recently, his research focus has been on electrocatalytic conversion of CO2 to useful building blocks for the chemical industry or energy sector.


