

Palladium-Catalyzed Silylcyanation of Ynamides: Regio- and Stereoselective Access to Tetrasubstituted 3-Silyl-2-Aminoacrylonitriles
Abstract
The palladium-catalyzed silylcyanation of ynamides is described. This reaction is fully regioselective, delivering tetrasubstituted 2-aminoacrylonitriles derivatives exclusively. Unexpectedly, the nature (aryl or alkyl) of the substituent located at the β-position of the ynamide directly controls the stereoselectivity. The reaction tolerates a number of functional groups and can be considered as the first general access to fully substituted 2-aminoacrylonitriles. Given the singular reactivity observed, a computational study was performed to shed light on the mechanism of this intriguing transformation. Relying on the specific reactivity of the newly installed vinylsilane functionality, the scope of 2-aminoacrylonitriles has been enlarged by postfunctionalization.
Introduction
Acrylonitriles have long been recognized for their versatility in synthetic chemistry and they are broadly used in various fields such as pharmaceutical or material sciences.1 In spite of a great need for such compounds, the methods available for obtaining fully substituted alkenyl nitriles remain limited. This can be easily explained by the inherent difficulty to synthesize tetrasubstituted alkenes because of the strong congestion around the C=C bond, and also the difficulty to distribute the various substituents in a stereo- and regioselective way.2 In addition, these difficulties are significantly increased when it is a question of having four substituents of a different nature around the alkene. In line with our interest in developing selective synthetic pathways towards amino-substituted Michael acceptors,3 we decided to turn our attention to the synthesis of tetrasubstituted 2-aminoacrylonitriles (also called α-enaminonitriles). Despite their high synthetic potential as captodative small and versatile nitrogen building blocks,4 and unlike β-enaminonitriles (i.e. 3-aminoacrylonitriles) for which many synthetic routes have been devised,5 2-amino-acrylonitriles remain underexplored. Moreover, no general pathways to reach tetrasubstituted members of this family have been described to date.
While very efficient, the condensation of aminonitrile derivatives with ketones6 or the nucleophilic substitution by an amino group of tetracyanoethylene,7 or a haloacrylonitrile,8 lead mostly to α-enaminonitriles exhibiting at least two identical substituents. One of the most elegant methods to rapidly build tetrasubstituted alkenes bearing four different substituents is the 1,2-difunctionalization of unsymmetrical internal alkynes.9, 10, 11 The first synthesis of tetrasubstituted 2-aminoacrylonitriles based on this approach was reported by the Beletskaya group.12 It consists of the silyl-, the stannyl- or the bromo-cyanation of ynamines catalyzed by trimethylsilyl iodide (Figure 1). However, this method is very limited in terms of substrates due to their restricted synthetic accessibility and their instability.13 The 1,2-difunctionalization strategy has also been applied by Werz and co-workers for the intramolecular chalcogenocyanation of two ynamides, leading to a fully substituted 3-sulfenyl-2-amido-acrylonitrile and a 3-selenyl-2-amido-acrylonitrile.14

Our strategy to access tetrasubstituted 2-aminoacrylonitriles and its scientific context.
In this context, we have decided to develop the regio- and stereo-controlled silylcyanation of internal ynamides15 (an easy-to-access family of nitrogen-substituted alkynes) to obtain fully substituted 3-silyl-2-amido-acrylonitriles 2. Interestingly, the introduction of a silylated moiety could increase the versatility of these platforms, given the rich reactivity of vinylsilanes.16 Thus, this transformation could allow to easily obtain a highly functionalized small building-block being at the same time an acrylonitrile, an enamide and a vinylsilane.
To date, very few methodologies allow for the alkyne silylcyanation. Besides Beletsakaya's work on ynamines presented above and including two examples of silylcyanation, only two groups have reported this type of transformation. The first study, by Chatani's group, focuses on the palladium-catalyzed silylcyanation of terminal alkynes mainly substituted by aryl groups. However, its application to an internal alkyne involved only diphenylacetylene as substrate and led to a complex mixture including traces of the targeted silylated acrylonitrile.17 The second work, reported by Ito and co-workers, is the palladium-catalyzed intramolecular silylcyanation of alkynes from a chlorosilane intermediately converted to a silyl cyanide. This reaction is stereoselective and regiospecific.18
Results and Discussion
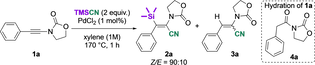
We first turned our attention to ynamide 1 a, bearing an oxazolidinone and a phenyl group (Table 1). This substrate is readily available, robust and exhibits an increased reactivity due to its push-pull electronic nature. In light of Beletskaya's work on ynamines,12 we tried to perform the silylcyanation reaction by means of Lewis acid catalysis. Unfortunately, whatever the acid used (see Supporting Information for details), the reaction systematically led to the hydration of the substrate to give amide 4 a. In direct relation to Chatani's report on the palladium-catalyzed silylcyanation of terminal alkynes,17 our best reaction conditions were defined as follows: 1 equivalent of ynamide (1 M in xylene), 2 equivalents of TMSCN and 1 mol% of PdCl2 at 170 °C for 1 h under argon atmosphere19 (Entry 1). Gratifyingly, under these conditions, the targeted product 2 a was obtained in 96 % yield as a sole regioisomer and with a Z/E ratio of 9 : 1. The configuration of the C=C bond of the major product, confirmed by X-Ray analysis (Figure 2), was determined as Z (i.e. resulting from an anti-addition of TMSCN onto the alkyne). This result is particularly interesting as it is opposite to the syn-addition selectivity observed by Chatani and co-workers. Moreover, the Z/E selectivity remained virtually unchanged (≈9 : 1 ratio), whatever the conditions tested during the optimization process. A control experiment showed that no reaction occurs in the absence of the catalyst (Entry 2). The use of less than 2 equivalents of TMSCN led to the partial conversion of the starting alkyne (Entry 3). In addition, a lower concentration of 1 a (Entry 4) or the increase of the catalyst loading (Entry 5) encouraged the formation of the hydrocyanation side product 3 a, thus inducing a decrease of the yield of the desired product 2 a. This side reaction was also promoted by decreasing the temperature (Entry 6). Quite remarkably, the use of a Pd0 catalyst such as Pd(PPh3)4 also led to the desired product 2 a in similar yield (Entry 7).20 Of note, only aromatic solvents such as xylene or mesitylene proved to be suitable for this transformation, while the other solvents tested led either to a complex mixture or to significantly lower yields of 2 a (Entries 8 and 9). Finally, other catalytic systems based on metals known to activate silylated compounds such as Ni0 or RhI have also been tested without any success (low yields or no conversion; Entries 10 and 11 respectively).
|
||||
Entry |
Deviations |
Conv. [%][b] |
Ratio[b] 2 a : 3 a |
2 a [%][c] |
|---|---|---|---|---|
1 |
None |
100 |
100 : 0 |
96 |
2 |
Without PdCl2 |
0 |
– |
– |
3 |
1 equiv. TMSCN |
55 |
100 : 0 |
51 |
4 |
Concentration=0.1 M |
100 |
65 : 35 |
55 |
5 |
Loading PdCl2=4 mol % |
100 |
86 : 14 |
73 |
6 |
Reaction at 130 °C |
100 |
60 : 40 |
33 |
7 |
Pd(PPh3)4 instead of PdCl2[d] |
100 |
100 : 0 |
92 |
8 |
DMF instead of xylene |
100 |
– |
0[e] |
9 |
Sulfolane instead of xylene |
100 |
100 : 0 |
46 |
10 |
Ni[P(OEt)3]4 instead of PdCl2 |
100 |
85 : 15 |
21 |
11 |
[Rh(COD)Cl]2 instead of PdCl2 |
0 |
– |
– |
- [a] See the Supporting Information for details. [b] Determined by 1H NMR. [c] Isolated yield. [d] Similar results could be obtained with other Pd0 catalytic systems, see Supporting Information for more details. [e] A complex mixture was obtained.

Scope of the silylcyanation of ynamides and X-ray analyses21 of the major isomers of 2 a and 2 al.
With a reliable set of conditions in hand, we then explored the scope and limitations of our methodology (Figure 2). First, keeping the oxazolidinone as the nitrogen moiety, we studied the effect of the introduction of various substituents at the aromatic ring. In general, all substitutions in para and meta positions led to very good results in terms of yields and stereoselectivity with Z/E ratios of about 9 : 1 (2 b–2 h). However, with compounds displaying an electron-withdrawing group at the aromatic ring (such a -CF3 2 e or a methyl ester 2 f), the yields were moderate due to the instability of the products formed, as witnessed during the purification process. The introduction of a methyl group at the ortho position (2 i), which implies a significant increase of steric hindrance, led to a lower 47 % yield and a lower stereoselectivity (Z/E=72 : 28). This detrimental influence of the steric demand on the stereoselectivity and efficiency of the reaction could be confirmed by comparing two positional naphthyl-derived isomers. In the case of a 3-naphthyl substituted ynamide, the reaction appeared as selective and nearly as efficient as with a phenyl group (2 j, 71 %, Z/E=91 : 9). Conversely, the 2-naphthyl substituted isomer furnished 2 k in only 36 % yield with a Z/E ratio of 60 : 40. Alkenyl-substituted ynamides proved to be relevant substrates in this transformation. While the substitution by a cinnamyl group led to the desired product 2 l in a moderate 45 % yield with a modest 65 : 35 Z/E selectivity, the substrate bearing a cyclohexenyl could be efficiently converted into product 2 m in 61 % yield with an excellent 95:05 Z/E selectivity. Additionally, heterocyclic substitutions of the alkyne such as thiophene, furan or pyridine proved to be compatible with this transformation leading to the corresponding silylcyanation products (2 n–2 r) in moderate to good yields (38–65 %) with Z/E ratios from 82 : 18 to 93 : 7. Then, we evaluated the effect of the modification of the nitrogen part of the ynamide. We first wanted to see if the steric hindrance on this side of the alkyne was as influential on the course of the reaction as it is on the other side. For this purpose, an ynamide bearing an oxazolidinone substituted by a phenyl group on one side and a phenyl group on the other was tested. In this case, the yield for the desired product 2 s was very good (83 %) but no stereoselectivity was observed. This result unambiguously demonstrated that the reaction is relatively sensitive to steric hindrance on both sides of the alkyne.
We then studied the tolerance of the reaction towards various electron-withdrawing groups. Pleasingly, most of them were found to be compatible with this silylcyanation reaction in terms of yield and stereoselectivity. Thus, substrates bearing lactams (2 t–2 u), sulfonamides (2 v–2 y) or acyclic carbamates (2 z–2 ae) all gave good results with yields generally close to 60–70 % and a Z/E selectivity around 80 : 20.
There were a few exceptions where the selectivity was logically lower due to the presence of more bulky groups on the nitrogen such as a cyclohexyl (2 aa), a benzyl (2 ab) or a phenyl (2 ac). It should be noted that the presence of an allyl group (2 ad), a Boc group (2 ae) or a phosphonate (2 af) on the nitrogen led to an important to complete degradation of the starting material under our reaction conditions. Finally, a substrate bearing an indole as the nitrogen moiety of the ynamide was efficiently transformed into the desired product 2 ag in 72 % yield with a Z/E ratio of 80 : 20.
Once the study on aryl- or alkenyl-substituted ynamides was completed, we turned our attention to alkyl-substituted substrates (2 ah–2 at). The reaction was as efficient and selective with these starting products but, unexpectedly, the stereoselectivity was systematically inversed in comparison with the previous series. As confirmed by the X-ray analysis of 2 al (Figure 2), this time, the major product, identified as (E)-2 al, was resulting from the syn-addition of TMSCN to the alkyne. On the other hand, the cyclopropyl-substituted ynamide 1 ao and the terminal ynamide 1 aq did not lead to any stereoselectivity. However, introduction of a more hindered nitrogen group such as a phenyl-substituted oxazolidinone restored it, leading to products 2 ap and 2 ar with Z/E ratios around 30 : 70. These results show once again the importance of the steric hindrance in the selectivity of this transformation. To finish this study on the possible variations of the reaction partners, we used different silylated groups. Thanks to an approach based on Patil's method22 to synthesize silylcyanides, we were able to obtain dimethylphenylsilyl cyanide (DMPS-CN) that, to make the sequence as safe as possible, was not isolated and the resulting solution, after filtration to remove all solids, was directly added to the silylcyanation reaction medium. Nicely, under these conditions and starting from ynamide 1 a, product 2 au bearing a DMPS group was obtained in 61 % yield with a Z/E ratio of 73 : 27.
To shed light on the mechanism of this transformation, we then performed some additional experiments (Figure 3). First, the configurational stability of the products was demonstrated by resubmitting the minor isomers of representative examples (namely E-2 a, Z-2 an, Z-2 aq and E-2 aq) to the reaction conditions without any isomerization after 1 h. Then, based on Chang's recent research on ynamide hydrosilylation23 and considering the sensitivity of our transformation to steric hindrance, we hypothesized that its key intermediate might be a silylated keteniminium which could be favored thanks to a β-silicon effect stabilization. To investigate this hypothesis, we placed ynamide 1 a in the presence of 1 equivalent of Pd(CN)2 and 2 equivalents of the electrophilic silicon reagent TMSCl, both potentially arising from the reaction of TMSCN with PdCl2, at 170 °C for 10 minutes. Interestingly, while the conversion was complete, only the hydrochlorination product 5 a and the hydration product 4 a were obtained in 57 % and 43 % yields respectively.24 The question raised by this first result was therefore whether Pd(CN)2 could indeed be a catalyst for this transformation. Positively, using Pd(CN)2 as catalyst, ynamide 1 a was converted into 2 a in 10 minutes with an isolated yield of 60 % and a Z/E ratio of 90 : 10, which is similar to what was observed with PdCl2. Finally, hypothesizing that the first step of the mechanism could be a carbopalladation involving Pd(CN)2, which could also explain the formation of the competitive hydrocyanation product which would then potentially arise from a protodemetalation of the formed intermediate, we performed the reaction without TMSCN in presence of a stoichiometric amount of this palladium complex. In this case, the conversion was just 33 % and only the hydration product 4 a was obtained without traces of the hydrocyanated product 3 a suggesting that this organometallic species is not involved in the formation of this competitive product.

Control experiments towards the elucidation of the mechanism.
In front of these various observations and the difficulties to draw strong conclusions about the course of the reaction, DFT computations at the M06L/def2-TZVPP level were performed to get a deeper insight into the mechanism (see the Supporting Information for computational details and the full study). The main results are presented in Figures 4 and 5. The numbering of the computed species follows that used in the Supporting Information. It is well-established that TMSCN preferentially gives isocyanide rather than cyanide complexes with transition metals or main group elements.25 Our study confirmed that the isocyanide palladium complex, shown below as an adduct with ynamide 1 a (complex G) is indeed the most stable form obtained from Pd(CN)2 and TMSCN (see also Scheme S1). The computed pathway leading to the minor product E-2 a is shown in Figure 4. The formation of the vinyl carbocation H from G is achieved through TSGH, lying at 20.8 kcal mol−1 on the free energy surface. The nucleophilic addition of the isocyanide provides the syn carbopalladation complex I, located at −2.1 kcal mol−1. The free energy of the corresponding transition state, TSHI, is 33.8 kcal mol−1. Manifold efforts failed to model a concerted carbopalladation. The question of the mechanism of the transformation of I into the final products E and E-2 a was then addressed. While a direct cleavage of the Pd−C bond by TMSCl could not be computed, we could model a shift of Me3Si+ to Pd to give species K, which precedes the silyldemetallation. This shift is yet not direct, it goes first through species J in which the silylium ion has moved towards a cyanide ligand. The highest-lying transition state of these three steps is TSKL, located at 36.4 kcal mol−1. This high value indicates that the cleavage of the Pd−C bond is slower than the carbopalladation, and is thus rate-determining. From the putative alkene-Pd complex L, the regeneration of E and liberation of compound E-2 a is a markedly exergonic step, placing the products at −19.7 kcal mol−1 on the energy surface.

Free energy profile (ΔG443, kcal mol−1) of the formation of the minor compound E-2 a by syn carbopalladation (selected distances in Å).

Free energy profile (ΔG443, kcal mol−1) of the formation of the major compound Z-2 a by anti carbopalladation (selected distances in Å).
We then studied the formation of the major compound Z-2 a, starting from M, which is preorganized for a trans attack of TMSNC (Figure 5). Complex M has virtually the same free energy as H (15.7 vs 15.5 kcal mol−1). The nucleophilic addition transition state TSMN lies at 25.4 kcal mol−1 on the free energy surface. The vinylpalladium complex N is more stable than the reactants by 2.2 kcal mol−1. We found only one way to cleave the Pd−C bond intramolecularly, and it does not require a Pd−Si intermediate in this case. It involves a shift of the Me3Si+ ion to the oxazolidinone carbonyl to give O at −2.0 kcal mol−1. A direct connection between O and the alkene complex P could be established through TSOP, located at 34.4 kcal mol−1. Such a carbonyl relay could not be modeled in the syn case. From P, addition of TMSNC to reform E and provide Z-2 a is strongly exergonic, the products lying at −21.1 kcal mol−1. We can notice that even in the anti case, this step is the highest-lying (34.4 kcal mol−1 vs 25.8 kcal mol−1 for the carbopalladation highest-lying step).
By comparing Figures 4 and 5, it is clear that both the carbopalladation and the cleavage of the Pd−C bond are favored in the anti case, which means that Z-2 a is expected to be the major product, as eventually observed experimentally. The E/Z-2 a experimental ratio is actually 1 : 9, which means a ΔΔG≠ of 1.935 kcal mol−1 between the rate determining steps at 443.15 K. The free energy difference between TSKL and TSOP is 2.0 kcal mol−1, meaning a predicted 1 : 9.7 between E-2 a and Z-2 a, which is close from the experimental value. The proposed mechanism also rationalizes why, in spite of a low difference in energy, E-2 a and Z-2 a are not equilibrated because the barrier on the way back cannot be overcome. The Z/E selectivity switch between aryl and alkyl ynamides has been studied and is reported in the Supporting Information. It transpires from this study that the stereoselectivity is dictated by the silyldemetallation, which is itself governed by steric factors. Lastly, the difficulty to achieve the cleavage of the Pd−C bond by the silylium ion is consistent with the occurrence of a competitive process, i.e. the protodemetallation leading to hydrocyanation side products from adventitious water or other proton sources.
After having demonstrated the feasibility of the silylcyanation of ynamides at the multigram scale (2 g of 1 a gave 71 % of pure Z-2 a isomer) and to illustrate the synthetic possibilities offered by this new family of synthons, we carried out several transformations taking advantage of the specific reactivity of the nitrile group or of the vinylsilane moiety (Figure 6). It should be pointed out that these transformations occurred with retention of the geometry of the C=C bond. First, protodesilylation by the action of cesium fluoride in methanol provided product 6 a quantitatively.12 Then, vinylsilane (Z)-2 a could be activated by a fluoride source such as TBAF to react as a masked vinyl anion to perform a nucleophilic addition to benzaldehyde to form the corresponding allyl alcohol 7 a in a good yield of 76 %.26 Cross-coupling reactions involving a vinylsilane exhibiting a TMS group being rather limited, we converted the Si-group into an iodine atom offering more versatility. For this, we found that iodine monochloride in dichloromethane as the only reaction condition providing vinyl iodide 8 a without isomerization in 90 % yield.27 The resulting product was then submitted to a Sonogashira-type coupling to afford the enyne 9 a in 51 % yield28 with a slight erosion of the Z/E ratio (81 : 19), while a Rosenmund-von Braun coupling afforded the dicyanated compound 10 a in 77 % yield29 with complete stereoretention. Finally, we studied the possibility of converting the nitrile group into another functionality, the difficulty being the presence of the TMS group, often considered to be sensitive under acidic conditions. Nevertheless, a modified Ritter reaction catalyzed by sulfuric acid in solution in t-BuOAc led to amide 11 a in 86 % yield.30

Gram-scale reaction on 1 a and follow-up chemistry on Z-2 a.
Conclusion
In conclusion, we have demonstrated that silylcyanation of ynamides is an efficient and versatile method for the synthesis of tetrasubstituted 2-aminoacrylonitriles. This palladium-catalyzed reaction is fully regioselective and its stereoselectivity appears to depend on the nature of the alkyne substitution of the ynamide with a stereodivergence between the aryl and alkyl groups. Computations support a mechanism involving first the formation of a keteniminium palladium complex (Pd−C bond formation), followed by addition of trimethylsilylisocyanide providing a vinylpalladium species (C−C bond formation). Cleavage of the Pd−C bond by the Me3Si+ then forms the Si−C bond, delivering the product scaffold. Ligand exchange between the product and trimethylsilylisocyanide then finishes the catalytic cycle. The opportunity of a fast and efficient diversification towards other molecules of interest was then demonstrated thanks to various post-functionalization taking advantage of the different functionalities present around these new building blocks.
Acknowledgements
P.H. is grateful to the French Ministry of Higher Education, Research and Innovation for a PhD fellowship. The authors thank the CNRS and the University of Strasbourg for financial support. M.D. is grateful to the French National Agency of Research (ANR) for the grant allocated to this project (C-Sil, ANR-CE07-19-0016). Dr. Trevor A. Hamlin (Free Uni. of Amsterdam) is thanked for fruitful discussions. Dr. Emeric Wasielewski (Uni. Strasbourg) is acknowledged for helpful discussions on NMR experiments. We thank Dr. Lydia Karmazin (Uni. Strasbourg) for X-ray analyses. V.G. thanks the UPSaclay, CNRS and Ecole polytechnique for financial support of this work. This work was granted access to the HPC resources of CINES under the allocation 2020-A0070810977 made by GENCI.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.

