

Fragment-Bindung an die Kinase-Scharnier-Region: Wenn Ladungsverteilung und lokale pKa-Verschiebungen etablierte Bioisosterie-Konzepte fehlleiten
Abstract
Die medizinisch-chemische Optimierung folgt einer Strategie, funktionelle Gruppen zu ersetzen und größere Substituenten an ein vielversprechendes Leitstrukturgerüst anzufügen. Wohl etablierte Bioisosterie-Regeln werden berücksichtigt, dennoch ist es schwierig abzuschätzen, ob die durchgeführten Modifikationen auch wirklich den Anforderungen einer Bindestelle gerecht werden. Die Elektronendichteverteilung und die pKa-Werte der Liganden werden moduliert und beeinflussen so Protonierungszustände und Bioverfügbarkeiten. Unter Berücksichtigung des benachbarten H-Brücken-Donor/Akzeptor-Musters des Scharnierbindungsmotivs (“Hinge-Region”) einer Kinase untersuchten wir kristallographisch eine Reihe von Fragmenten, um zu sehen, ob sie das erforderliche Interaktionsmuster erwidern können. Unerwarteterweise sind Benzoesäure und Benzamidin, mit den richtigen Substituenten dekoriert, ebenso wie ein Carboxamid oder eine phenolische OH-Gruppe völlig bioisoster. Ein einzähniger Pyridinstickstoff übertrifft sogar zweizähnige Funktionalitäten am Liganden. Die Bedeutung der korrekten Einstellung von pKa-Werten der angefügten funktionellen Gruppen am Liganden durch zusätzliche Substituenten am Molekülgerüst wird damit offensichtlich.
Die Leitstrukturoptimierung in der medizinischen Chemie folgt in der Regel einer Strategie, bei der ein vielversprechendes Grundgerüst einer Leitstruktur modifiziert wird, indem funktionelle Gruppen ersetzt und größere Substituenten und Molekülteile angefügt werden, so dass ein Bindungsverhalten unverändert erhalten bleibt. Dabei werden etablierte Regeln der Bioisosterie berücksichtigt.1-5 Heutzutage fließen meist Strukturdaten des Zielproteins mit in das Design ein und Dekorationen des Grundgerüsts werden hinsichtlich des sterischen Anspruchs und einer Wechselwirkungs-Komplementarität mit der Proteinbindestelle ausgewählt. Auch dieser Schritt ist wieder von Bioisosterie-Überlegungen geprägt und kann durch Modellierungssoftware unterstützt werden. Ebenso werden Proteine mit ähnlichen Taschen konsultiert, um über die dort bindenden Liganden Ideen für das weitere Design zu erlangen.6, 7 Die stetig wachsende Zahl an Kristallstrukturen, die in der öffentlich zugänglichen PDB-Datenbank hinterlegt sind, verleiht solchen Strategien das erforderliche Potential.8
Es ist jedoch äußerst schwierig abzuschätzen, ob die gewählten Auswechselungen von funktionellen Gruppen oder Gerüsterweiterungen ideal zu den Eigenschaften der vorgegebenen Bindungsstelle passen. Die Beurteilung dieser Auswahl betrifft Eigenschaften wie die korrekte Platzierung einer geladenen oder stark polarisierten Gruppe, die die Gesamtverteilung der Elektronendichte über die angefügte Gruppe beeinflusst oder die Einstellung eines optimalen pKa-Wertes bestimmt. Dies legt die Protonierung des Liganden fest und beeinflusst dessen Bioverfügbarkeit.9-12 Darüber hinaus wird auch das Grundgerüst, an das die bioisostere Austauschgruppe chemisch gebunden wird, je nach der Chemie zum Erstellen der Verknüpfung einen Einfluss nehmen. Diese Einflüsse können sterischer wie elektronischer Natur sein. Letztere werden die Elektronendichteverteilung im Molekül in Abhängigkeit von der gewählten Verknüpfungschemie abwandeln. Folglich können pKa-Werte oder die Platzierung und Direktionalität von Wechselwirkungen verändert werden.13 Daher ist es kaum möglich exakt vorherzusagen, ob z. B. die erforderlichen pKa-Eigenschaften der zu ersetzenden funktionellen Gruppe optimal zu den Wechselwirkungen passen, die von der Gegengruppe des Proteins verlangt werden. Dazu ist es wahrscheinlich, dass die Proteinumgebung Effekte induziert, die die Elektronendichte umverteilen und den Liganden zusätzlich polarisieren.14 Infolgedessen müssen Medizinische Chemiker oft feststellen, dass der geplante bioisostere Ersatz nicht die erwartete Affinitätssteigerung in den neu entworfenen und mühsam synthetisierten Zielmolekülen erreicht.
Hier wären die folgende Information sehr wertvoll: Wie würde die für den Ersatz geplante Gruppe an das Protein binden, wenn sie frei von räumlichen und elektronischen Zwängen durch die Bindung an das Grundgerüst der Leitstruktur mit dem Protein interagieren könnte? So würde das optimale Interaktionsmuster mit der Proteinumgebung offensichtlich. Eine Möglichkeit könnte die Verwendung von ausgeklügelten Modellrechnungen sein, um diese Frage zu beantworten. Da es sich jedoch um Verschiebungen der Elektronendichte, Polarisationseffekte und in der Folge um Modulationen von Protonierungszuständen handelt, werden nur quantenchemische Berechnungen auf hohem Niveau aussagekräftige Antworten liefern.15-19 Diese sind derzeit noch weit von einer Routineanwendung auf Proteine entfernt. Es erfordert langjährige Erfahrung in der adäquaten Verwendung solcher Berechnungen, um schlüssige Informationen zu erhalten. Darüber hinaus benötigen sie enorme Rechenkapazitäten. Als Alternative könnte daher, zumindest solange solche dringend benötigten Werkzeuge fehlen, auf experimentelle Daten zurückgegriffen werden.
Wir haben kürzlich aus Neutronenbeugungsversuchen gelernt, die experimentell zuverlässige Informationen über Wasserstoffatome in Protein-Ligand-Komplexen liefern, dass die Aminogruppe des Anilins protoniert wird, sobald das Molekül in der S1-Tasche der Serinprotease Trypsin gebunden wird. Diese Tasche beherbergt an ihrem tiefvergrabenen Ende eine negativ geladene Carboxylatgruppe eines Aspartatrests. Formal entspricht die Protonierung von Anilin einer Verschiebung des pKa-Werts von Anilin (pKa=4,6) in Wasser um vier Größenordnungen wenn man berücksichtigt, dass die Kristallisation bei einem pH=7,5 vorgenommen wurde. Überraschenderweise beträgt der gegenseitige Abstand zwischen der Ammoniumgruppe des Anilins und der Carboxylatgruppe des Proteins 4 Å.20
Da dieses Beispiel zeigt, dass langreichende Polarisationseffekte schwer vorherzusagen sind und die Entwicklung geeigneter Rechenmodelle experimentelle Referenzinformation benötigt, beschlossen wir, einen experimentellen Ansatz zu verfolgen. Er sollte Daten über die Modulation der Bindungseigenschaften von funktionellen Gruppen sammeln, von denen erwartet wird, dass sie bioisostere Eigenschaften aufweisen. Wir wollten sehen, wie sich solche Gruppen an der Proteinbindestelle verhalten, wenn keine räumliche Beschränkung durch die Bindung an das Grundgerüst eines größeren Liganden vorliegt. Für unsere Studie wählten wir die Scharnierregion einer Kinase und untersuchten eine Reihe niedermolekularer Sonden, auch “Fragmente” genannt,21 im Hinblick auf ihre angenommenen Bindepositionen an dem Kinase-Hinge-Motiv. Wir berücksichtigten insbesondere die pKa-Eigenschaften und die Modulation der Elektronendichteverteilung über das Grundgerüst in Abhängigkeit der angefügten Substituenten.
In den letzten 30 Jahren wurde ein weites Spektrum chemischer Strukturbausteine entdeckt, die das Erkennungsmuster komplementieren, das durch die Aminosäuren der Kinase-Hinge-Region vorgegeben wird.22, 23 Es wurden ein-, zwei- und dreizähnige Interaktionsmuster vorgeschlagen, die mit dem Peptidstrang der Scharnierregion wechselwirken können.24 Beobachtete Unterschiede in diesen Motiven wurden ausgenutzt, um nicht nur die Affinität, sondern insbesondere die Selektivität für einzelne Mitglieder des Kinoms zu optimieren.25-27
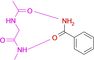
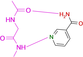
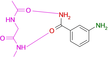
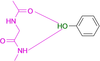
Wir wählten die gut kristallisierende und umfassend untersuchte cAMP-abhängige Proteinkinase A (PKA) aus.28 Sie erkennt den Adeninteil des natürlichen Substrats ATP und nutzt benachbarte Donor- und Akzeptorfunktionalitäten der zugänglichen Peptidbindungen der Scharnierregion, um den Kofaktor über Wasserstoffbrücken zu binden.29 Ein ideales Pendant zur Ergänzung des vorgegebenen Donor-/Akzeptormusters der Hinge-Region wäre daher eine Carboxamidgruppe im Liganden. Wir begannen daher mit Benzamid als Fragmentsonde. Der eingenommene Bindungsmodus an der Hinge-Region weist das erwartete H-Brückenbindungsmuster zweier parallel ausgerichteter Donor/Akzeptorfunktionen mit der Scharnierregion auf. Zu unserer Überraschung ändert sich jedoch durch die Einführung eines pyridinähnlichen Stickstoffs im Phenylring die Bindungspose völlig, und das Fragment zieht es vor, seinen Pyridinstickstoff zur Interaktion mit der Scharnierregion zu verwenden. Die Anlagerung einer Aminogruppe als elektronenschiebendem Substituent zeigt wiederum die erwartete Bindungspose. Aber schon die Einführung einer Hydroxylgruppe verändert die Bindungsgeometrie und ein Teil der Moleküle interagiert über die Hydroxylgruppe mit der Hinge-Region.
Diese recht überraschenden Ergebnisse, die mit sehr einfachen Liganden erzielt wurden, haben uns dazu angeregt, unsere Studie auf eine Vielzahl anderer funktioneller Gruppen, einschließlich Benzoesäuren und Benzamidine, auszudehnen. Völlig unerwartet ergaben sich eine Benzoesäure und ein Benzamidin, wenn sie mit den richtigen Substituenten versehen sind, als zwanglos bioisoster, vorausgesetzt der korrekte Protonierungszustand stellt sich ein.
Es gelang uns, die Kristallstrukturen von 19 Fragmenten mit einem zentralen aromatischen Ring und daran angefügter Substituenten mit abweichenden Eigenschaften zu bestimmen (Tabelle 1). Ihre pKa-Werte wurden in wässriger Lösung vermessen. Alle Fragmente binden im aktiven Zentrum neben der Scharnierregion, wo sonst der natürliche Ligand ATP gebunden wird. Die einzelnen Bindungsmodi von 1–19 sind in Tabelle 1 schematisch skizziert und in Abbildung 1, Abbildung 2 und Abbildung 3 dargestellt. Eine detaillierte Beschreibung der einzelnen Bindungsmodi mit den verschiedenen Interaktionsmustern ist in den Supporting Information zusammen mit dem zur Bestimmung der pKa-Werte angewendeten Protokoll beschrieben.

Überblick über die verschiedenen Liganden, wie sie an die Scharnierregion der PKA binden. A: Benzamid 1 (PDB-Code: 6SNX), B: Pyridin-3-carboxamid 2 (5N3H), C: 3-Aminobenzamid 3 (5N3Q), D: 4-Hydroxybenzamid 4 (5N3S), E: Phenol 5 (6Z44), F: 3-Amino-5-(trifluormethyl)-1H-pyridin-2-on 6 (5N33). Hier und in den folgenden Bildern sind die Heteroatome dem Typ entsprechend kodiert (N: blau, O: rot, F: türkis, S: gelb, Wassermoleküle: rote Kugeln, H-Bindungen als gestrichelte Linien mit Abständen in Å. Kohlenstoffatome sind für die verschiedenen Komplexe unterschiedlich gefärbt).

Überblick über die verschiedenen Liganden, wie sie an die Scharnierregion der PKA binden. A: 4-(Trifluormethyl)benzamid 7 (6SPS), B: 6-(Morpholin-4-yl)pyridin-3-carboxamid 8 (6SPY), C: Terephthalsäuremonoamid/ 4-Carbamoylbenzoesäure 9 (6SOX), D: Benzoesäure 10 (6SNN), E: 4-(Trifluormethyl)benzoesäure 11 (6SPM), F: 3-Aminobenzoesäure 12 (6SPU).

Überblick über die verschiedenen Liganden, wie sie an die Scharnierregion der PKA binden. A: 4-Nitrobenzoesäure 13 (5N3J), B: 4-Nitrophenol 14 (6Z08), C: 6-Dimethylaminopyridin-3-carbonsäure 15 (5N3E), D: 4-(Trifluormethyl)benzcarboximidamid 16 (5N3D), E: 4-Hydroxybenzamidin 17 (6YPS), F: Thiophen-3-carboximidamid 18 (5N3C), G: Iso-Nicotinamidin 19 (6ZN0).
No. |
Chemische Formel (Scharnierregion links, violett) |
pKa- Werte |
PDB- Code |
Auflösung [Å] |
|---|---|---|---|---|
1 |
|
<2 >12 |
6SNX |
1.40 |
|
|
|
|
|
2 |
|
<2 3.37 >12 |
5N3H |
1.36 |
|
|
|
|
|
3 |
|
<2 3.68 >12 |
5N3Q |
1.31 |
|
|
|
|
|
4 |
|
<2 8.36 >12 |
5N3S[a] |
1.14 |
|
|
|
|
|
5 |
|
9.99[b] |
6Z44 |
1.38 |
|
|
|
|
|
6 |
|
1.33 10.43 |
5N33 |
1.43 |
|
|
|
|
|
7 |
|
<2 >12 |
6SPS |
1.65 |
|
|
|
|
|
8 |
|
<2 3.74 >12 |
6SPY |
1.60 |
|
|
|
|
|
9 |
|
<2 3.53 >12 |
6SOX |
1.38 |
|
|
|
|
|
10 |
|
4.01 |
6SNN |
1.82 |
|
|
|
|
|
11 |
|
3.48 |
6SPM |
1.37 |
|
|
|
|
|
12 |
|
3.17 4.56 |
6SPU |
1.39 |
|
|
|
|
|
13 |
|
3.20 |
5N3J |
1.12 |
|
|
|
|
|
14 |
|
6.91 |
6Z08 |
1.49 |
|
|
|
|
|
15 |
|
3.05 6.17 |
5N3E |
1.53 |
|
|
|
|
|
16 |
|
10.78 |
5N3D |
1.77 |
|
|
|
|
|
17 |
|
7.75 >12 |
6YPS |
1.35 |
|
|
|
|
|
18 |
|
11.32 |
5N3C |
1.77 |
|
|
|
|
|
19 |
|
2.23 10.12 |
6ZN0 |
1.59 |

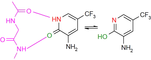
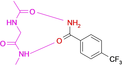
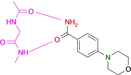
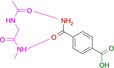
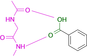


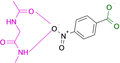
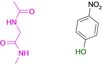
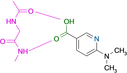
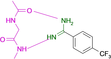
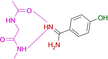
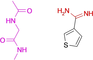
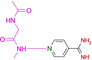
- [a] Zweite Ausrichtung mit Carboxamid-Gruppe zur Scharnierregion. [b] Wert aus dem CRC-Handbuch für Chemie und Physik.30
Bei Kinasen stellt die Scharnierregion ein typisches Erkennungsmuster bereit, an das das natürliche Substrat ATP bindet. Da sich die Arzneimittelentwicklung auf die Hemmung der ATP-Bindungsstelle konzentriert hat, müssen vermeintliche Leitstruktur-Kandidaten funktionelle Gruppen enthalten, die auf dieses Muster reagieren können. Die Scharnierregion gibt die Amidbindungen des Peptidrückgrats vor, an die über Wasserstoffbrückenbindungen der mutmaßliche niedermolekulare Ligand bindet. Daher scheint ein alternativ ausgerichtetes Muster von H-Brücken-Donor- und Akzeptor-Funktionen auf den ersten Blick ideal zu sein, um die Eigenschaften des Proteins zu erwidern.
Um die Bindungseigenschaften niedermolekularer Liganden mit den gewünschten Eigenschaften zu untersuchen, wählten wir kleine aromatische Fragmente aus, die eine Carboxamid-Funktion an oder eingebettet in das aromatische Ringsystem aufweisen. Erwartungsgemäß weist ein großer Teil der untersuchten Fragmente genau das erwartete Muster von zwei nahezu parallelen Wasserstoffbrückenbindungen zu der Scharnierregion auf (vgl. Fragmente 1, 3, 4, 7, 8, 9, Abbildung 1 und Abbildung 2). Ziemlich überraschend ist die Bindungsposition von 2, da dieses Fragment mit seinem pyridinähnlichen Stickstoff an die Scharnierregion bindet und die Carboxamidgruppe in ein Netzwerk von überbrückenden Wassermolekülen eingebettet ist. Der Stickstoff vom Pyridintyp besitzt einen pKa-Wert von 3,37 in wässriger Lösung und ist offensichtlich der bessere Akzeptor für eine H-Brücke, die von der Scharnierregion ausgeht. Unwahrscheinlich ist, dass das in Wassermoleküle eingebettete Carboxamid der bestimmende Faktor ist, der zu dem veränderten Bindungsmodus führt.
Eine zweite Überraschung ist Fragment 4, das in der Kristallstruktur mit zwei verschiedenen Orientierungen bindet, entweder über die erwartete Wechselwirkung mit der Carboxamidgruppe, oder aber überraschenderweise über seine Hydroxylfunktion, die sich am entgegengesetzten Ende des Fragments befindet. Die OH-Gruppe (pKa=8,36) scheint in der Lage zu sein, sowohl als H-Brückendonor wie auch als Akzeptor für die Scharnierregion zu dienen. Dies wird durch den Bindungsmodus des Phenols 5 bestätigt (Abbildung 1 E). Selbst die Einbettung der Carboxamid-Funktion in den sechsgliedrigen Ring von 6 erlaubt die erwartete zweizähnige Wechselwirkung mit der Scharnierregion. Sie erfordert jedoch das Tautomer mit aufgebrochener Aromatizität. Dazu wird der pyridinartige Stickstoff (pKa=1,33) protoniert und fungiert als H-Brückendonor für den Carbonylsauerstoff des Scharniers.
Interessanterweise können auch Benzoesäurederivate als Bindungspartner für die Scharnierregion dienen, sofern sie im neutralen Zustand das erforderliche H-Brücken-Donor/Akzeptor-Muster annehmen (10, 11, 12, 15, Abbildung 2 und Abbildung 3). In diesem Protonierungszustand bilden sie ein Bindungsmuster aus, das der Carboxamidgruppe sehr ähnlich ist. Die Bindungsgeometrien der Benzoesäure 10 (Abbildung 2 D) und Benzamid 1 (Abbildung 1 A) sind praktisch identisch. In wässriger Lösung liegen die pKa-Werte der hier untersuchten Säuren in einem Bereich zwischen 3–4, so dass die neutrale Form in wässriger Lösung auch die wahrscheinlichste Form in der Bindungstasche ist.
Überraschenderweise fällt die p-Nitrobenzoesäure 13 (Abbildung 3 A) völlig aus diesem Muster heraus. Obwohl ihr pKa-Wert im gleichen Bereich liegt (3,20), zieht es das Fragment vor, mit der Scharnierregion über seine Nitrogruppe nur einzähnig zu interagieren. Auf den ersten Blick scheint dies eher ungewöhnlich, da die Nitrogruppe bekanntlich ein schlechterer H-Brückenakzeptor ist als die Carbonsäure. Dennoch sind eine ganze Reihe von Protein-Ligand-Komplexen bekannt, bei denen die Nitrogruppe an einer H-Brücke beteiligt ist. In der PDB konnten 506 Wechselwirkungen dieser Art gefunden werden (Details siehe SI), wobei neben 13 ein weiterer Ligand über eine H-Brücke an die Scharnierregion einer Kinase bindet (PDB-Code: 2W7X). Im Komplex mit 13 wird die gegenüberliegende Carboxylatgruppe in eine Position gebracht, in der sie eine ladungsunterstützte Wechselwirkung mit der Ammoniumgruppe von Lys72 eingehen kann. Wir gehen daher von einer Deprotonierung der Säuregruppe aus. Damit wird die Ausbildung einer Salzbrücke zu Lys72 die treibende Kraft, um den Protonierungszustand dieser Gruppe in den deprotonierten Zustand zu verschieben. Es entsteht eine energetisch begünstigte Salzbrücke. Basierend auf diesen Erfahrungen haben wir Nitrophenol 14 getestet (Abbildung 3 B). Es weist die Nitrogruppe auf, es fehlt aber eine gegenüberliegende Funktion, die leicht deprotoniert werden kann (pKa=6,91), um einen ladungsunterstützten Kontakt zu Lys72 herzustellen. Infolgedessen vermeidet 14 jeglichen Kontakt mit der Scharnierregion, ganz anders als 4 und 5. Stattdessen platziert 14 seine Nitrogruppe neben Lys72, erreicht dadurch aber nur eine sehr lange Wasserstoffbrücke mit kaum idealer Geometrie.
Das vielleicht überraschendste Ergebnis stellt der Bindungsmodus des Benzamidinderivats 16 dar (Abbildung 3 D). Aromatische Amidine sind die typischen Gruppen, die aufgrund ihrer hohen pKa-Werte, die weit über 12 liegen, häufig zur Interaktion mit Asp oder Glu in Proteinen verwendet werden. Liganden, die mit elektronenziehenden Gruppen am aromatischen Teil ausgestattet sind, können jedoch wie 16 niedrigere pKa-Werte (hier 10,78) aufweisen. Dies ermöglicht dem Molekül, mit einer ungeladenen Amidinofunktion an die Scharnierregion zu binden und dann das erforderliche H-Brücken-Donor/Akzeptor-Muster aufzuweisen. In diesem Zustand nimmt das Benzamidinfragment den gleichen Bindungsmodus an wie die Benzoesäurederivate (z. B. 10–12, 15), ein mehr als überraschender und definitiv unerwarteter bioisosterer Ersatz! Eine Analyse der in der PDB verfügbaren Strukturen zeigt, dass alle an ein aromatisches Ringsystem gebundenen Amidinogruppen als Wasserstoffbrückendonor fungieren, wenn sie an ein Protein gebunden werden (für Details siehe SI).
Diese Beobachtung regte uns an, weitere aromatische Amidine zu untersuchen. Die Fragmente 17 und 18 weisen höhere pKa-Werte für die Amidinofunktion auf. 17 (Abbildung 3 E) kontaktiert die Scharnierregion einzähnig, sehr ähnlich wie die p-Nitrobenzoesäure 13. Fragment 18 vermeidet es völlig, mit dem Scharnier zu interagieren. Stattdessen rekrutiert es die Carboxylatgruppe von Asp184, um eine Salzbrücke zu bilden. Wir nehmen an, dass dies die treibende Kraft für die Bildung dieses Bindungsmodus darstellt.
Ein interessanter Fall ist wiederum Fragment 19, da es eine Amidinogruppe aufweist, die noch weniger basisch ist als die in 16 (pKa(19)=10,12; pKa(16)=10,78). Dennoch kontaktiert 19 das Scharnier nicht über ein H-Brücken-Donor/Akzeptor-Muster wie 16, sondern verwendet seinen pyridinähnlichen Stickstoff (pKa=2,23), um eine H-Brücke von der Scharnierregion aufzunehmen, ähnlich wie Fragment 2. Hier ist der angenommene Protonierungszustand der Amidinogruppe schwer abzuschätzen. Da die Gruppe jedoch nur drei ziemlich lange H-Brücken ausbildet, erscheint der ungeladene Zustand sehr wahrscheinlich. Wie bei 2 ist das bestimmende Merkmal für den eingenommenen Bindungsmodus von 19 die über den pyridinartigen Stickstoff eingegangene Wechselwirkung. Im Gegensatz dazu verfügt Fragment 16, das einen ähnlichen pKa-Wert besitzt, nur über die Amidinogruppe zur Bildung von H-Brücken. Die Präferenz von 19 für die Wechselwirkung des pyridinartigen Stickstoffs mit der Scharnierregion stimmt mit der für 2 gefundenen Situation überein. Interessanterweise führten mehrere Arzneimittelentwicklungen dazu, dass ein Stickstoff vom Pyridintyp als einzige Wechselwirkungsgruppe zur Bindung an die Scharnierregion verwendet wird (z. B. Lapatinib, Gefitinib, Erlotinib, Vandetinib, Tanduinib, SB202190, SB203580).31, 32 Möglicherweise ist ein einzelner Kontakt zur Interaktion mit der Scharnierregion vorteilhaft im Vergleich zu einer überfrachteten mehrfachen Substitution mit nahe beieinander stehenden funktionellen Gruppen am Liganden. Es sollte nicht vergessen werden, dass ein zweizähniges H-Brückenmuster aus Donor- und Akzeptorfunktionen auf gegenüberliegenden Seiten der interagierenden Moleküle immer zu abstoßenden sekundären Wechselwirkungen führt, die die ausgebildeten Wasserstoffbrücken schwächt. Diese Beobachtung wurde erstmals von Zimmerman et al.33 auf dem Gebiet der Wirt-Gast-Chemie beschrieben. Es spielt jedoch auch bei Protein-Ligand-Interaktionen eine Rolle, wie wir vor einiger Zeit für die Ligandbindung an ein tRNA-modifizierendes Enzym zeigen konnten.34
Zusammenfassend lässt sich sagen, dass die vorliegende Studie die Merkmale der ATP-Bindetasche der bekannten Kinase PKA ausleuchtet, indem sie für eine Reihe von Fragmenten die Wechselwirkungen mit der Scharnierregion des Enzyms untersucht. Das Peptidrückgrat der Hinge-Region stellt ein Muster von H-Brücken-Donor/Akzeptor-Funktionen bereit, das von einem potentiellen Liganden erwidert werden muss. Daher erscheinen Liganden, die eine Gruppierung von nahe beieinander liegenden Donor- und Akzeptorgruppen aufweisen auf den ersten Blick ideal. Funktionelle Gruppen wie z. B. ein Carboxamid, eine Carbonsäure, aber auch eine Amidinogruppe im ungeladenen Zustand erfüllen das geforderte Muster. Das verbleibende Molekül muss lediglich mit geeigneten elektronenschiebenden oder -ziehenden Gruppen dekoriert werden, um den erforderlichen Protonierungszustand zu gewährleisten. Ein solches Design erfordert jedoch eine exakte Kontrolle der pKa-Eigenschaften eines Moleküls. Aufgrund von Polarisationseffekten in der Bindungstasche können pKa-Werte, die in wässriger Lösung definiert sind, leicht um mehrere Größenordnungen verschoben werden (vgl. den oben erwähnten Trypsin-Anilin-Fall20). Dies macht solche Vorhersagen extrem schwierig. Darüber hinaus beobachteten wir, dass einige überraschende Bindungsmodi resultieren, die besondere Eigenschaften einer Bindungstasche ausnutzen. So können funktionelle Gruppen, die hinsichtlich ihrer pKa-Werte ähnlich sind, dennoch mit unterschiedlichen Protonierungszuständen in einem anderen lokalen Bereich der Tasche binden (z. B. 13, 18, 19). Insbesondere ein pyridinähnlicher Stickstoff im Liganden kann zu überraschenden Änderungen des Bindungsmodus führen, was sich vermutlich durch die Bevorzugung einer einfachen Wasserstoffbrücke zur Hinge-Region unter der Vermeidung abstoßender sekundärer Wechselwirkungen erklären lässt. Diese treten aber zwangsläufig bei gegenüberliegenden zweizähnigen Wasserstoffbrücken-Kontakten unvermeidbar auf.
Hinterlegungs-Codes: PKA-Ligand-Komplexe (ID, PDB-Code): Benzamid (1, 6SNX), Pyridin-3-carboxamid (2, 5N3 H), 3-Aminobenzamid (3, 5N3Q), 4-Hydroxybenzamid (4, 5N3S), Phenol (5, 6Z44), 3-Amino-5-(trifluormethyl)-1H-pyridin-2-on (6, 5N33), 4-(Trifluormethyl)benzamid (7, 6SPS), 6-(Morpholin-4-yl)pyridin-3-carboxamid (8, 6SPY), 4-Carbamoylbenzoesäure (9, 6SOX), Benzoesäure (10, 6SNN), 4-(Trifluormethyl)benzoesäure (11, 6SPM), 3-Aminobenzoesäure (12, 6SPU), 4-Nitrobenzoesäure (13, 5N3J), 4-Nitrophenol (14, 6Z08), 6-Dimethylaminopyridin-3-carbonsäure (15, 5N3E), 4-(Trifluormethyl)benzcarboximidamid (16, 5N3D), 4-Hydroxybenzamidin (17, 6YPS), Thiophen-3-carboximidamid (18, 5N3C), Iso-Nicotinamidin (19, 6ZN0).
Acknowledgements
Wir möchten uns besonders für die Hilfe und Unterstützung durch die Beamline-Wissenschaftler bei der Sammlung der Beugungsdaten am BESSY II (HZB, Berlin, Deutschland), DESY, P11 (Hamburg, Deutschland) und ESRF, ID29 (ESRF, Grenoble, Frankreich) bedanken. Dem HZB danken wir für finanzielle Unterstützung bei den Reisekosten. Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Conflict of interest
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.

