Natural history and genotype-phenotype correlations in 72 individuals with SATB2-associated syndrome
Abstract
SATB2-associated syndrome (SAS) is an autosomal dominant disorder characterized by significant neurodevelopmental disabilities with limited to absent speech, behavioral issues, and craniofacial anomalies. Previous studies have largely been restricted to case reports and small series without in-depth phenotypic characterization or genotype-phenotype correlations. Seventy two study participants were identified as part of the SAS clinical registry. Individuals with a molecularly confirmed diagnosis of SAS were referred after clinical diagnostic testing. In this series we present the most comprehensive phenotypic and genotypic characterization of SAS to date, including prevalence of each clinical feature, neurodevelopmental milestones, and when available, patient management. We confirm that the most distinctive features are neurodevelopmental delay with invariably severely limited speech, abnormalities of the palate (cleft or high-arched), dental anomalies (crowding, macrodontia, abnormal shape), and behavioral issues with or without bone or brain anomalies. This comprehensive clinical characterization will help clinicians with the diagnosis, counseling and management of SAS and help provide families with anticipatory guidance.
1 INTRODUCTION
SATB2-associated syndrome (SAS; Glass syndrome, OMIM 612313) is a multisystemic disorder first described in 1989 in a 16-year-old male with an interstitial deletion of 2q32.2-2q33.1, severe intellectual disability, cleft palate, short stature, and craniofacial dysmorphism (Glass, Swindlehurst, Aitken, McCrea, & Boyd, 1989). With the subsequently discovered link between SATB2 and the development of cleft palate, several additional individuals who had alterations in SATB2 as a result of contiguous deletions, intragenic deletions and duplications, and translocations were described (Zarate & Fish, 2017). Similarly, following the first report of a SATB2 single nucleotide mutation in a 36-year-old man with cleft palate, generalized osteoporosis, and profound intellectual disability, several additional individuals have been described with coding mutations (Bengani et al., 2017; Leoyklang et al., 2007; Zarate et al., 2015; Zarate, Kalsner, et al., 2017). Consistent features described in individuals with SAS, regardless of the molecular mechanism, include neurodevelopmental disabilities, behavioral abnormalities, and craniofacial anomalies (Zarate & Fish, 2017; Zarate, Kaylor, & Fish, 1993). From the neurodevelopmental perspective, all individuals described thus far have had developmental delay (DD)/intellectual disability (ID) with severely limited speech (Bengani et al., 2017; Zarate & Fish, 2017; Zarate, Kalsner, et al., 2017). Behavioral issues may include hyperactivity, sleeping difficulties, autistic features, obsessive tendencies and/or aggressiveness (Zarate, Kalsner, et al., 2017). Common craniofacial anomalies include palatal abnormalities, micrognathia, and abnormal dentition, most commonly crowding or abnormal dental size/shape (Zarate, Kalsner, et al., 2017). Other reported features include decreased bone density, hypotonia, growth retardation, and epilepsy (Zarate & Fish, 2017). While common dysmorphic features have been described in individuals with SAS, facial appearance has not been thought to be distinctive enough to clinically diagnose SAS (Bengani et al., 2017; Zarate, Kalsner, et al., 2017).
Despite advances in the phenotypic characterization of SAS, longitudinal data in SAS patients have not been available. Furthermore, the vast majority of published reports have been limited to children. Here we report information from the largest number of individuals with SAS and across a broad age range. We provide the phenotype, genotype, and natural history of 72 individuals enrolled through the SAS clinical registry, 46 of whom are not previously published.
2 MATERIALS AND METHODS
Participants were recruited into the SAS clinical registry through a referral by a treating clinician, a facilitated inquiry by the testing laboratory, direct contact by a caregiver, or via the SAS support group. Individuals with a molecularly confirmed diagnosis of SAS were eligible for the study. All families reported herein agreed to share clinical information and were enrolled under a research clinical registry protocol approved by the Institutional Review Board of the University of Arkansas for Medical Sciences. For all individuals, medical records including laboratory results were reviewed. Supplementary information was also obtained by parental report through a REDCap™ questionnaire. All families who shared photographs signed consent for publication. Individuals SATB2-01, 03, 04, 05, 06, 07, 10, 14, 16, 18, 20, 22, 23, 24, 27, 28, 31, 32, 33, 35, 36, 47, 58, 59, 60, and 67 were included in previous publications (Bengani et al., 2017; Docker et al., 2014; Schwartz, Wilkens, Noon, Krantz, & Wu, 2017; Tomaszewska et al., 2013; Zarate et al., 2015; Zarate, Kalsner, et al., 2017; Zarate, Steinraths, et al., 2017).
Analysis and comparison of 2D photographs was conducted using Face2Gene (FDNA, Inc., Boston, MA), an analytic tool that automatically detects and evaluates subtle craniofacial dysmorphology in order to identify a recognizable facial pattern for rare syndromes based on 2D facial photos (Basel-Vanagaite et al., 2016).
During the study period, different chromosomal microarray platforms were used to analyze genomic DNA extracted from peripheral blood. Earlier patients had testing using oligonucleotide-based platforms (Agilent Human Genome 44 K and 180 K Oligo array, Agilent Technologies, Santa Clara, CA) while more recent cases were evaluated using the CytoscanHD whole genome SNP array (Affymetrix, Santa Clara, CA). Whenever possible, confirmation of microarray findings were performed by FISH analysis. In other patients, clinically available next generation sequencing (NGS) techniques including whole exome sequencing (WES) were utilized. Sequence variants were confirmed by PCR or Sanger sequencing. In silico prediction tools for interpretation of the variants reported in this study included SIFT (http://sift.jcvi.org/), MutationTaster (http://www.mutationtaster.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), CADD (http://cadd.gs.washington.edu/), and Provean (http://provean.jcvi.org/index.php). Pathogenicity of missense variants was ascribed based on the American College of Medical Genetics and Genomics (ACMG) variant interpretation guidelines (Richards et al., 2015).
2.1 Statistical analysis
For genotype/phenotype correlations, individuals’ demographic and clinical milestones were characterized with medians and means according to the genetic alteration type (large deletion, intragenic deletion, disruptive coding variant, missense variant). Large deletions were defined as those larger than 1 Mb as determined by molecular cytogenetic testing. Group medians/means were compared with the Wilcoxon–Mann–Whitney or t-test. Phenotype frequencies sorted by genetic alteration type groups were analyzed with the binomial homogeneity test for multiple responses (Ho: proportion in phenotype groups are the same across genetic alteration type). Analyses were carried out in SAS 9.4 (SAS Institute, Cary, NC).
3 RESULTS
Seventy-two individuals were recruited to the study from around the world (Supplementary Figure S1a), comprising 41 males and 31 females. The mean age of recruited individuals was 8.8 years (range: 3 months to 34 years, Supplementary Figure S1b) while the mean age at diagnosis was 6.6 years (range: 1 week to 34 years). A variety of genetic alterations resulted in SATB2 disruption were found (Supplementary Figure S1c). Cumulative prevalence of clinical features within this cohort is summarized in Figure 1a.

3.1 Perinatal history
In agreement with previous reports, the mean gestational age, weight, and length at birth were normal at 39 weeks (range 32–42 weeks), 3.28 kg (range 1.34–4.53 kg), and 50.5 cm (range 41.5–58.4 cm), respectively.
3.2 Growth and nutrition
Feeding difficulties during infancy were reported in 72% (51/71) of individuals (Figure 1b). Those difficulties persisted past infancy in 47% (33/70) with six needing gastrostomy tubes. Thirty-nine percent (28/72) of the cohort experienced postnatal growth problems, although mostly during infancy. Sialorrhea was identified in 83% (57/69) and Botulinum toxin injections of salivary glands were needed for three individuals with some improvement.
3.3 Vision
Visual problems have been previously reported in SAS. Zarate, Kalsner, et al. (2017) Strabismus was reported in 36% (25/70) and refractive errors in 21% (15/70) of individuals.
3.4 Cardiovascular, renal, respiratory, and immunology
Congenital heart defects were identified in two individuals (3%), and both resolved spontaneously: a patent ductus arteriosus with pulmonary artery stenosis in one and an atrial septal aneurysm in another. A duplicated ureterocele with hydronephrosis and isolated hydronephrosis were diagnosed in two individuals, both needing surgical repair. Isolated instances of hypospadias and hydrocele were seen. Although immune problems are not a feature of SAS, a single individual was suspected to have Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections (PANDAS). Recurrent ear infections were reported in a few individuals while restrictive lung disease and asthma were infrequent.
3.5 Endocrine
A single individual was diagnosed with type 1 diabetes while two had hypothyroidism. Although it is unclear how many participants were tested, low vitamin D was diagnosed in five individuals, all of whom were treated.
3.6 Craniofacial and dental anomalies
Palatal and dental abnormalities are consistent features of SAS. In addition to cleft palate (40%), high-arched palate (25%) and bifid uvula (3%) were also present. Micrognathia was present in 46% of affected individuals but none required surgical correction. Dental anomalies (more commonly crowding, large incisors, or abnormal dental shape) were present in all individuals after the first year of life (Figure 1d).
3.7 Skeletal
Skeletal abnormalities such as pectus excavatum, scoliosis, tibial bowing, and abnormal bone mineralization have been reported occasionally (Boone et al., 2016; Lee et al., 2016; Rainger et al., 2014; Talkowski et al., 2012; Tegay et al., 2009; Zarate et al., 2015; Zarate, Kalsner, et al., 2017) and reaffirmed in this study (Figure 1e). The phenotype and genotype description of seven individuals with SAS documented to have low bone mineral density (BMD) and included in this cohort was recently reported (Zarate, Steinraths, et al., 2017). The high frequency of abnormal mineralization was further supported by six additional individuals who were documented to have a slightly low (−1.9 to −1.1SD) or low (<−2SD for age) BMD by energy X-ray absorptiometry (DXA) scans for 76% (13/17) of those scanned (Supplementary Table S1). Similarly, of the 18 individuals who had serum alkaline phosphatase levels performed, 12 (67%, 8 females) were found to be elevated (Figure 1f).
3.8 Neurological
Abnormal brain imaging has been reported in several individuals with SAS (Zarate & Fish, 2017; Zarate, Kalsner, et al., 2017). Magnetic resonance imaging (MRI) of the brain had been performed for 55 individuals in our cohort at a mean age of 2.8 years (range 1–7 years); abnormalities were detected in 47% (26/55) (Figure 1c). White matter abnormalities were the most common finding (29%, 16/55), typically described as hyperintense T2/FLAIR white matter signals in the periventricular region. Delayed myelination and enlarged cerebral ventricles were also seen. While these abnormal neuroradiographic findings are common, they are not pathognomonic and do not appear to be prognostic. Seizure disorders, gait abnormalities, and hypotonia have been reported previously (Bengani et al., 2017; Zarate, Kalsner, et al., 2017) and were present in 19, 37, and 74% of our cohort, respectively. Twenty-nine additional individuals underwent electroencephalograms (EEG) with abnormalities detected in 7 (slow background, abnormal wakefulness, or epileptiform discharges), but none were categorized as clinical seizures. For those with seizures, there is no predominant seizure type, and a variety of antiepileptic medications have been used with variable success alone or in combination: levetiracetam (four individuals), valproic acid (two individuals), and single instances of oxcarbazepine, carbamazepine, and lamotrigine use. Dystonia was diagnosed in two females (3%) at 8 and 11 years of age, respectively. The former was managed with trihexyphenidyl first, followed by carbidopa/levodopa with improvement, the latter with no significant improvements after trials of carbidopa/levodopa and baclofen.
3.9 Neurodevelopment
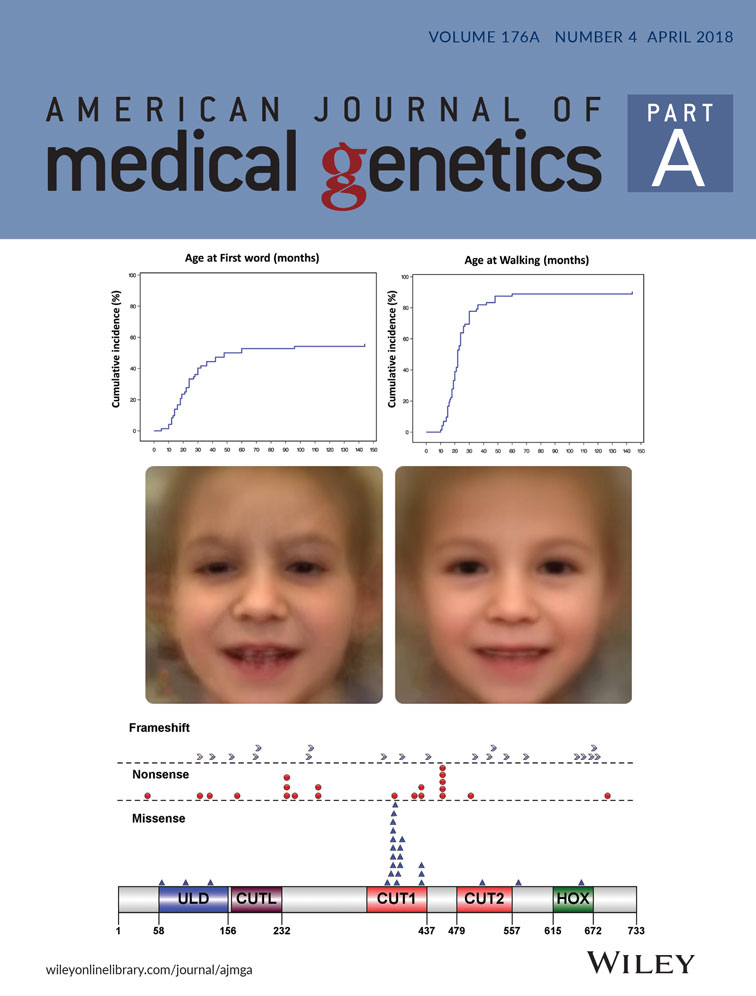
Developmental delay/ID is a hallmark feature of SAS (Bengani et al., 2017; Zarate et al., 2015; Zarate & Fish, 2017; Zarate, Kalsner, et al., 2017). Key developmental milestones were typically delayed for those old enough to have been assessed (Figure 2a–c). Severe delay in speech acquisition was universal (median at first word 24 months, range 10–144 months), and 82% of individuals have limited vocabulary of less than 10 words or absent speech by 10 years of age (Figure 2d). Twelve individuals had cognitive assessments performed. Composite nonverbal and full scale cognitive scores were reported in seven (range: 30–62, mean 48.2 ± 11) and eight (range: 32–53, mean 45.5 ± 6.9) individuals, respectively.

3.10 Behavioral phenotype
A broad spectrum of behavioral findings is observed in SAS individuals (Zarate, Kalsner, et al., 2017). Individuals are often described as having a jovial or friendly personality. In childhood, behavior is commonly described as difficult with episodes of frustration, tantrums, and meltdowns. They can become agitated or have aggressive outbursts (Figure 2e). These aggressive tendencies are often physical, directed towards self or others, and typically reported in teenagers or adults. Four individuals (ages 10.5, 14, 18, and 32 years) were on risperidone. Three had good response, while the fourth needed several additional psychotropic medications over his lifetime to achieve control (valproic acid, haloperidol, sulpiride, quetiapine, chlorprothixene, and fluvoxamine). The potential complexity and worsening of the behavioral issues with age was further illustrated by two additional older individuals (20 and 34 years) who required antipsychotic medications (olanzapine and floropipamide). Sleeping difficulties have been managed with clonidine in some individuals.
3.11 Facial phenotype
Non-specific craniofacial dysmorphisms have been described in individuals with SAS (Bengani et al., 2017; Zarate, Kalsner, et al., 2017). In this study, at least minor facial dysmorphic features were described in 47 individuals (65%) (Figure 3). The most commonly identified facial dysmorphisms included thin vermilion of the upper lip, flat philtrum, prominent chin, deeply set eyes, micrognathia and abnormal dentition. Progressive coarsening of facial features was appreciated in older individuals (Figure 3k–m). For additional objectivity, a total of 100 2D photographs from 56 individuals (39 with intragenic pathogenic variants, 12 with large deletions, 3 with intragenic microdeletions, and 2 with microduplications) were analyzed using automated facial analysis (Figure 3n). Automated facial analysis (gestalt) by the Face2Gene tool vs.17.6.1, without other phenotypic information, suggested SAS as number 1 or at least within the top 10 or top 30 considerations automatically in 7%, 27%, or 51% of individuals, respectively.

3.12 Genotype and inheritance
3.12.1 Cytogenetic abnormalities
Of the 26 individuals diagnosed with cytogenetic abnormalities that included SATB2, 16 had large deletions that encompassed the entire SATB2 gene as well as other adjacent genes, 8 had small intragenic deletions, and 2 had small microduplications (Figures 4a and 4b; Supplementary Table S2). The latter two cases were siblings who were documented to have a 2.9-Mb microduplication that was not present in either parent, suggestive of gonadal mosaicism. These siblings displayed characteristic phenotypic features (neurodevelopmental delay with profound speech delay, dental anomalies, and distinctive facial features) and with their duplication predicted to disrupt SATB2. Of the remaining 24 individuals, parental studies were performed in 7 individuals and were de novo in each case.

3.12.2 Intragenic pathogenic variants
Identification of the SATB2 coding variants were made by WES (39/46, 85%) or NGS panels (7/46, 15%). When trio analysis was conducted (39 families, including a pair of monozygotic twins), the pathogenic change was found to be de novo in 97% (38/39). In one case, a c.1174G>C (p.G392R) variant was also found in the blood sample of her unaffected father at a low level (not quantified) through WES and Sanger sequencing data. Missense pathogenic variants were the most frequent type of coding pathogenic variant (88% de novo) and mainly located in the CUT1 domain (Figure 4c, Supplementary Figure S1d, Supplementary Table S3). Further supporting evidence for interpretation of missense variants can be found in Supplementary Table S4.
4 DISCUSSION
SAS is a multi-system disorder caused by alterations of the SATB2 gene, a transcription factor that binds to nuclear matrix-attachment regions (MARs) and activates the transcription of genes that are critical to the development of multiple organ systems, tissues, including the jaw, brain, and skeleton (Britanova et al., 2006; Dobreva et al., 2006; Dobreva, Dambacher, & Grosschedl, 2003; Gyorgy, Szemes, de Juan Romero, Tarabykin, & Agoston, 2008; Jaitner et al., 2016). In this manuscript we provide the basis to better characterize the natural history and assess genotype-phenotype correlations. We anticipate that as sequencing technologies improve additional individuals with alterations in SATB2 will be identified.
4.1 Prevalence
While the prevalence of SAS is currently unknown, two recent studies have given an estimated frequency of SAS in large cohorts of individuals with undiagnosed intellectual disability/developmental delay of 0.24–0.3% (Bengani et al., 2017; Zarate, Kalsner, et al., 2017).
4.2 Diagnosis
SAS is generally difficult to recognize early in life. Although clinical criteria have not been established, analysis of the over 120 individuals reported thus far with SAS confirms that the main features of this syndrome include neurodevelopmental disabilities with severely limited speech, defects of the palate, a range of dental anomalies, osteopenia, and orthopedic issues, nonspecific brain MRI findings of delayed myelination and ventriculomegaly and behavioral issues that worsen with age. We suggest that SAS should be suspected in any child with this combination of features. While nonspecific brain MRI abnormalities are commonly seen and could support the consideration of SAS, their diagnostic value and clinical utility remains unknown.
Given the high frequency of deletions/duplications affecting the SATB2 gene, chromosomal microarray analysis is recommended as a first-tier test followed by a multi-gene panel (SATB2 is included in several clinically available panels targeting ID, seizures, or atypical Rett/Angelman syndrome) or comprehensive genomic sequencing (Zarate et al., 1993). Of importance, while almost all probands with SAS reported in the literature to date have a de novo genetic alteration, parental mosaicism can rarely be observed. Of the 65 families reported thus far with trio analysis, two instances of mosaicism have now been documented (4.6%): a previously reported case of gonadal mosaicism and the case here described of low level somatic mosaicism in a paternal blood sample (Bengani et al., 2017). We also describe another instance of presumed germline mosaicism for two siblings with identical microduplications not found in their parents.
4.3 Genotype–phenotype correlations
Given the limited number of reported individuals with SAS, no formal genotype–phenotype correlations have been established other than there being a higher frequency of genitourinary anomalies, cardiac defects, and ectodermal changes (other than dental) in individuals with large deletions involving SATB2 and adjacent genes (Zarate & Fish, 2017). In this study, we included individuals with large deletions given their overlapping phenotype (and sometimes indistinguishable) with those affected by SAS caused by other genetic alterations. With the exception of HECW2, other genes linked to neurodevelopmental findings that were deleted in some individuals in this study, have a recessive pattern of inheritance. Dominant coding mutations in HECW2 were recently reported to be associated with a neurodevelopmental disorder but haploinsufficiency has not been documented as the mechanism with functional studies (Berko et al., 2017). A comparison of demographic and phenotypic traits among individuals with different genetic alterations (large deletion, intragenic deletion, disruptive pathogenic variants, missense pathogenic variants) was conducted (Supplementary Tables S5 and S6). Individuals with large chromosomal deletions were diagnosed at earlier ages (mean 2.5 years, p ≤ 0.0006). Individuals with missense or disruptive pathogenic variants were more commonly reported to have sialorrhea (p = 0.0115), and those with large deletions were more likely to have a history of growth retardation (p = 0.0033). While no clear neurodevelopmental differences were documented by this comparison, an analysis of this and other phenotypic traits (low bone density, feeding difficulties, cleft palate) will need a larger sample size to document potential differences. Lastly, SAS does cause common facial dysmorphisms that allow facial recognition technology to suggest this diagnosis within the top 30 of possible diagnoses in over half of individuals. Individuals with coding pathogenic variants were more likely to have SAS suggested at least within the top 10 list (17/39 = 44%) compared to those with large deletions (3/12 = 25%) but this observation was not statistical significant (p = 0.32).
4.4 Management and surveillance
There is currently no specific treatment available for SAS and therefore, management is symptomatic. Surveillance guidelines have been previously proposed (Zarate et al., 1993; Zarate & Fish, 2017). Early in life, the emphasis should be placed on nutritional support for feeding difficulties and management by a cleft/craniofacial team when palatal anomalies are present. A referral for developmental support and special education instruction is needed. While ongoing speech therapy is often reported to be beneficial by families of individuals with SAS, progress can be slow or limited and there is currently no available prospective data to determine adequate frequency of services. Attention should also be placed on communication therapy focusing on facilitated and nonverbal communication strategies. Ongoing surveillance of growth abnormalities, ophthalmological and dental anomalies, and epilepsy is recommended. With the recent report of the high frequency of low BMD in individuals with SAS, evaluation for scoliosis/spine deformity with consideration of screening bone density after 5 years of age is appropriate. Some providers are starting to use inhibitors of bone resorption in the setting of low BMD and osteopenia, but more data will be needed to determine ideal management options (Zarate, Steinraths, et al., 2017). Current experience with the use of antiepileptic and psychotropic medications is anecdotal and no singular drug is proven to be more beneficial. We provide a brief summary of medications used in this cohort to date as a starting point while additional data are collected.
ACKNOWLEDGMENT
The authors are grateful to all participating families. Clinicians seeking further information or advice on SAS can consult the dedicated website (www.satb2gene.com) or contact the corresponding author. WK Chung received support from the Simons Foundation and the JPB Foundation.
CONFLICTS OF INTEREST
AA is an employee of FDNA, Inc. All other authors declare no conflicts of interest.