KBG syndrome: An Australian experience
Abstract
In 2011, heterozygous mutations in the ANKRD11 gene were identified in patients with KBG syndrome. Since then, 100 cases have been described with the expansion of the clinical phenotype. Here we present 18 KBG affected individuals from 13 unrelated families, 16 with pathogenic mutations in the ANKRD11 gene. Consistent features included intellectual disability, macrodontia, and the characteristic broad forehead with hypertelorism, and a prominent nasal bridge. Common features included hand anomalies, cryptorchidism, and a large number of palate abnormalities. Distinctive findings in this series included malrotation of the abdominal viscera, bilateral inguinal herniae in two patients, basal ganglia calcification and the finding of osteopenia in three patients. Nine novel heterozygous variants were found and the genotype-phenotype correlation was explored. This report highlights the need for thorough examination and investigation of the dental and skeletal systems. The results confirm the specificity of ANKRD11 mutations in KBG and further evidence for this transcription repressor in neural, cardiac, and skeletal development. The description of further cases of KBG syndrome is needed to further delineate this condition, in particular the specific neurological and behavioral phenotype.
1 INTRODUCTION
Facial dysmorphism, macrodontia, skeletal anomalies, and developmental delay have been identified as the major features of KBG syndrome (Brancati et al., 2004 ; Herrmann, Pallister, Tiddy, & Opitz, 1975 ). Clinical diagnostic criteria are based on a review of 50 patients (Skjei, Martin, & Slavotinek, 2007 ). Bone age examination in childhood was suggested to be removed from diagnostic criteria (Ockeloen et al., 2015 ).
In 2011, whole exome sequencing in two affected families identified mutations in ANKRD11 (Sirmaci et al., 2011 ). Sanger sequencing found de novo truncating ANKRD11 mutations in three other cases. Four patients had mutations in exon 10 and one family had a mutation at the exon 12 splice site acceptor site according to transcript [NM_001256182.1].
In a recent study, 19 reported patients had mutations in exon 10 of ANRKD11 gene (Ockeloen et al., 2015 ). ANKRD11 was initially hypothesised as a tumour suppressor gene. ANKRD11 overexpression inhibits transcriptional activation in vitro (Zhang et al., 2004 ). The transcriptional regulatory domains of the protein include two repressor domains at the N and C termini and an additional activation domain (Zhang, Li, & Chen, 2007 ). The ANKRD11 protein is highly expressed in the human brain and localized to nuclei of neurons and glial cells (Gallagher et al., 2015 ). ANKRD11 influences the expression of several genes related to neural development, highlighting its association with the neurobehavioral and developmental phenotype in KBG syndrome, and providing a mechanism for less common phenotypic associations such as periventricular nodular heterotopia (Oegema et al., 2010 ). While studies on peripheral blood mRNA from a patient with a truncating ANKRD11 mutation demonstrated incomplete nonsense mediated decay, a dominant negative effect was also hypothesized (Walz et al., 2015 ). Similarities between the 16q24.3 microdeletion syndrome and KBG syndrome suggest that these are overlapping entities mediated by ANKRD11 haploinsufficiency (Goldenberg et al., 2016 ; Willemsen et al., 2010 ). Results from the DDD triome study in the United Kingdom found that mutations in ANKRD11 accounted for around 1% of patients with an undiagnosed developmental delay (Wright et al., 2015 ). This finding reflects that KBG syndrome is still an under-recognized clinical condition. Discovery of patients with apparent non-syndromic intellectual disability with ANKRD11 mutations has led to revised diagnostic criteria for KBG syndrome (Low et al., 2016 ). These proposed criteria exclude costovertebral anomalies and delayed bone age, promoting otitis media, and hearing anomalies along with seizures, cryptorchidism, feeding problems, palate insufficiency, and delayed anterior fontanelle closure.
We report on 18 patients from 13 families with KBG syndrome to further expand the phenotypic and mutational spectrum.
2 METHODOLOGY
This study includes 18 patients from 13 families referred to genetic services in Australia between 2005 and 2016. Sixteen patients from 11 families had genetic testing. Two other patients who were diagnosed on the basis of clinical criteria did not have genetic testing. Ten patients were clinically diagnosed with KBG syndrome based on the Washington criteria by a clinical geneticist and had Sanger sequencing of ANKRD11. Six patients were diagnosed after testing of a customised gene panel for intellectual disability, whole exome sequencing, or whole genome sequencing.
Written informed consent was obtained for inclusion in the studies and the use of clinical reports and photographs in accordance with local ethics protocols. All patients underwent standard chromosomal microarray according to local laboratory procedures. Genomic sequences are reported with respect to transcript NM_001256182.1 [GRCh37]. In silico prediction of pathogenicity included AlignGVGD, Alamut visual, SIFT, and Polyphen. Variants predicted to cause truncation or to affect proper splicing were considered pathogenic.
We reviewed the previously reported individuals with proven mutations or deletions in the ANKRD11 gene and compared them with our patients.
3 CLINICAL REPORTS
The clinical, radiologic, and molecular findings are summarized in Tables 1-3 and Figures 1-4 , and in supplementary material.
| CASE | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| FAMILY | 1 | 1 | 1 | 2 | 3 | 3 | 3 | 4 | 5 |
| AU1.1 | AU1.2 | AU1.3 | AU2.1 | AU3.1 | AU3.2 | AU3.3 | AU4.1 | AU5.1 | |
| Gender | M | M | F | F | F | F | F | F | M |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Thai | Caucasian |
| Birth measurements, weight, length and OFC (in centiles) | [10-50th], [50th], [50th] | [50-90th], NK, [10-50th] | NK | [50th], [50-90th], [50-90th] | [< 10th] [<10th], [< 10th] | NK | NK | [10-50th], NK, [50th] | [10-50th] [50-90th [<10th] |
| Current Height (in centiles) | 25th | 3rd | 50th | 25-50th | 75th | 3rd | 3rd | 1st | 25th |
| Current OFC | 50th | 2-50th | 50-98th | 50th | 50th | 50th | 5-98th | NK | 50th |
| Perinatal Issues | pre-eclampsia | DCDA twin NICU-FTT | – | Antepartum haemorrhage | IUGR, feeding difficulty | - | NK | IUGR, conjugated hyperbilirubinemia | - |
| Craniofacial | High forehead Triangular face Short neck | Brachyturricephaly High forehead | Triangular face Short, webbed neck | Broad forehead Triangular face | Broad forehead | Brachycephaly Broad forehead Midface hypoplasia | Broad forehead | Triangular face | Triangular face Small, retrognathic chin |
| Dysmorphic Features | Ptosis Downslanting palpebral fissures Overfolded helix Low anterior hairline Thick vermillion Long flat philtrum | Downslanting palpebral fissures Anteverted nares High nasal bridge Externally rotated ears Patchy hair loss Thick vermillion Long philtrum | Downslanting palpebral fissures Hypertelorism High nasal bridge Anteverted, low set ears Low anterior hairline Synophrys Thick vermillion | High nasal bridge Bulbous nasal tip Flat left pinna Frontal upsweep Sparse hair Everted upper, thick vermillion Flat philtrum | Hypertelorism Left sided strabismus Anteverted nares High nasal bridge Thin nasal tip Dysplastic left ear Sparse hair Synophrys Thick vermillion Short philtrum | Hypertelorism High nasal bridge Thin nasal tip Simple ears Synophrys Thin vermillion Short philtrum | Ptosis Hypertelorism Thin vermillion Short philtrum | Upslanting palpebral fissures Hypertelorism Anteverted nares Bulbous nasal tip Low set ears, Pits on lobes Bushy brows Long philtrum | Hypertelorism Short upturned nose Anteverted nares High, broad, prominent nasal bridge Prominent ears Low anterior hairline Broad brows flared laterally Synophrys Long philtrum |
| Development | Severe language delay | Global DD Severe language delay | Learning difficulty | Global DD Severe language delay | Global DD language delay | In lower class at school | learning difficulties | Global DD | Global DD moderate to severe language delay |
| Neurological features | ADHD | Hypotonia Poor sleep MRI brain: small optic nerves Developmental venous anomaly left frontal lobe | Hyperreflexia Bipolar affective disorder | ADHD Anxiety | ADHD aggressive, inflexible, physical outbursts | – | – | MRI brain: low lying conus medullaris partial agenesis corpus callosum choroid plexus cyst | Seizure onset age 3 Seizures ceased age 6 Autism Spectrum Disorder ADHD Significant behavioural MRI brain − small arachnoid and pineal cysts |
| Vision | Normal | Retrolental fibrosis Grade IVb ROP [blind] | Myopia Astigmatism, Night blindness | Astigmatism. | Strabismus Refractive error | Normal | Normal | Hypermetropia, Glasses Corneal lesions | Refractive error |
| Hearing | Bilateral hearing loss NOS | – | – | – | – | Right sided hearing loss since birth | – | Severe mixed hearing loss and enlarged vestibular aqueducts | – |
| Palatal irregularity | high arched and narrow | bifid uvula | high arched | – | nasal speech, palate incompetent | nasal speech, palate incompetent | nasal speech, palate incompetent | – | Bifid uvula, slightly high palate |
| Dental Features | Macrodontia crowding premature loss of permanent teeth | Primary dentition | Macrodontia fused central incisors | Macrodontia | Macrodontia | Macrodontia premature loss of permanent teeth | Macrodontia premature loss of permanent teeth | primary dentition | Macrodontia malposition crowding |
| Other systemic features | Bilateral cryptorchidism | Ventricular septal defect and patent ductus arteriosus on echocardiogram | – | Recurrent intussusception Psoriasis | – | Hypothyroidism | Repaired ventricular septal defect and atrial septal defect | Ectopic kidneys Abdominal viscera malrotation and abnormal vessels, PEG fed | Normal echocardiogram Bilateral inguinal hernia repair Bilateral orchidopexy for undescended testes FTT, GORD from early infancy, settled by 6 months |
| Skeletal Features | Delayed bone maturation Normal survey | Delayed bone maturation Mild osteopenia Otherwise normal survey | Minor thoracolumbar scoliosis | Mild thoracic scoliosis Mild osteopenia | Normal skeletal Survey | Hypoplastic 12th rib | Degenerative lumbar spine, convex right scoliosis, hyperostosis frontalis interna | Bilateral coxa valga | Mild thoracic scoliosis convex right |
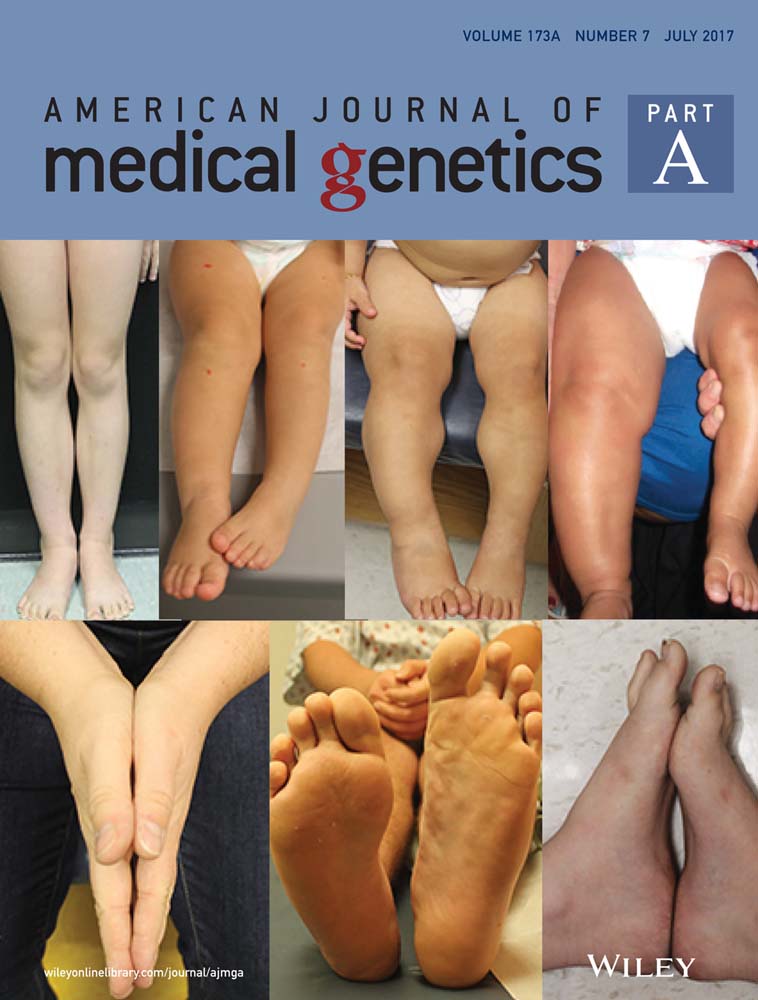
| Hand and foot anomalies | Bilateral cutaneous 2-3 finger and toe syndactyly | 5th finger brachydactyly | 5th finger contracture | Bilateral short 4th, 5th metacarpals | 5th finger clinodactyly | Brachydactyly 5th finger clinodactyly | Brachydactyly | fixed flexion 5th fingers, bilateral clinodactyly | Bilateral 2-3 and 3-4 cutaneous toe syndactyly |
| Meets clinical criteria | YES | YES | YES | YES | YES | YES | YES | YES | YES |
| CASE | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 |
|---|---|---|---|---|---|---|---|---|---|
| FAMILY | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 12 | 13 |
| AU6.1 | AU7.1 | AU8.1 | AU9.1 | AU10.1 | AU11.1 | AU12.1 | AU12.2 | AU13.1 | |
| Gender | M | F | F | F | M | M | F | M | F |
| Age at Assessment | 15 | 28 | 34 | 4 | 15 | 12 months | 6 | 33 | 4 and 6 |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Pacific Islander |
| Delivery | SVD, term, suctioning and oxygen | 38 weeks, SVD | NK | 40 weeks, SVD | SVD at 40 weeks, fetal distress and meconium liquor | 29 weeks, emergency LSCS | SVD at 40 weeks | NK | 32 weeks LSCS for failure to progress |
| Birth measurements, weight, length and OFC (in centiles) | [50th] [>50th [>90th] | [10th], NK, NK | NK | [10th], [10th], [3rd] | [<10th], [50th] [50th] | [25th], NK, [25th] | [<10th], NK, NK | NK | [90th],NK, [50th] |
| Current Height | 1st | 10th-25th | <3rd | 10th | Height <3rd and improved to 25th with growth hormone | Corrected for prematurity, 10th | 10-25th | 75th-90th | Height <3rd, no response to growth hormone |
| Current Head Circumference (OFC, in centiles) | 2-50th | 2nd | 2nd | 10th | 50th | 25th | 50th | 50th | 2-50th |
| Perinatal Issues | Difficulty feeding, reflux, hypotonia | Maternal smoking exposure Congenital bilateral hip dislocation | NK | - | Advanced maternal age Respiratory distress | IUGR, pre-eclampsia Jaundice patent ductus arteriosus TPN required | - | NK | Seizures |
| Dysmorphic Features | |||||||||
| Craniofacial | Round face | Brachycephaly | Pointed chin Short neck | Large anterior fontanelle with delayed closure Triangular face | Brachycephaly Triangular face, micrognathia Short neck | Large anterior fontanelle with delayed closure Turricephaly | Webbed neck | Broad forehead | Short neck |
| Dysmorphic Features | Upslanting palpebral fissures Hypertelorism Strabismus Prominent nasal bridge Hypoplastic alae nasi Low set, protruding ears Low hairline anteriorly Bushy eyebrows Micrognathia Thin upper vermillion | Horizontal Palpebral fissures Relative hypertelorism High nasal bridge Bulbous nasal tip Hypoplastic alae nasi Simple helices Wild hair Low hairline Broad eyebrows Synophrys | Upslanting palpebral fissures Hypertelorism Asymmetric nares High nasal bridge Bulbous nasal tip Hypoplastic alae nasi Anteverted dysplastic ears Bushy eyebrows Thick vermillion | Hypertelorism High nasal bridge Bulbous tip Dysplastic ears Pre-auricular pits Synophrys Thin vermillion Long philtrum | Epicanthic folds Hypertelorism Bulbous nasal tip Low set, posteriorly rotated ears, Left overfolded helix; Right anteverted Thick hair Low posterior hairline Broad/bushy eyebrows Thin vermillion Long, flat philtrum | Hypertelorism Anteverted nare Thin vermillion Long, flat philtrum | Long Palpebral fissures Ptosis Bilateral strabismus repair Anteverted nares Bulbous tip Low set, prominent ears Low hairline Broad/bushy eyebrows Thin vermillion | Low columella Simple helices | Long Palpebral fissures Microphthalmia Hypertelorism Prominent columella Bulbous tip Hypoplastic alae nasi Prominent ears Low hairline Broad/bushy eyebrows Synophrys Thin vermillion Flat philtrum |
| Developmental Hx | Gross motor delay Stutter Nocturnal enuresis mild-moderate intellectual disability | Mild intellectual disability | Mild global developmental delay | Mild global developmental delay IQ 70 | Mild DD Language delay | Borderline at 12 months corrected | Fine motor difficulty Language delay | No delay Learning difficulty at school | Expressive and receptive language delay Mostly non-verbal at 6, and signing |
| Neurological features | nil | moody, aggressive, difficult behaviour | nil | ADHD | ADHD | Hypotonia ADHD MRI brain and inner ear: dilated left endolymphatic sac; arachnoid cysts | Hypotonia | – | Neonatal seizures MRI brain: Bilateral T2 hyperintensities, Multiple calcific foci in brain, arachnoid cyst, germinolytic cysts |
| Vision | squint repair, refractive error and astigmatism | – | – | – | – | Fine horizontal nystagmus, Grade II ROP | Normal after strabismus repair | – | Astigmatism |
| Hearing | – | – | – | – | Conductive Hearing loss | – | Left sided conductive hearing loss | – | Bilateral moderate conductive hearing loss with hearing aids |
| Palatal irregularity | High arched palate | – | – | – | High palate | – | – | – | High palate |
| Dental Features | Macrodontia Crowding | Macrodontia Crowding Long dental roots on XR | Macrodontia Fused central incisors Malposition | Primary dentition | Macrodontia | Primary dentition Delayed eruption | Primary Dentition | 8 mm Central incisors Malposition Crowding | Primary teeth just lost |
| Additional systemic features | Primum atrial septal defect and cleft mitral valve GORD Recurrent ENT/respiratory infections | Bilateral inguinal hernia Blind sacral pit | - | Normal echocardiogram | Normal echocardiogram Severe cystic acne Recurrent otitis media | Bilateral inguinal herniae Bilateral cryptorchidism | Patent foramen ovale Malrotated bowel | ||
| Skeletal Features | Delayed bone maturation Pectus excavatum Increased lumbar lordosis Otherwise normal survey | Narrow spaces in thoracic spine Congenital bilateral hip dysplasia | Flattened vertebrae, end plate depression No Scoliosis Short phalanges | Skeletal survey not done as no apparent abnormalities | Delayed bone maturation Bilateral cervical ribs Wormian bones Inferior pectus excavatum Pes planus | Right clavicular pseudoarthrosis | Delayed bone maturation | Skeletal survey not done as no apparent abnormalities | T9 fracture, calcified T11-T12 disc osteopenia |
| Hand and foot anomalies | Brachydactyly | Short tubular bones in fingers | Hand and foot anomalies | Brachydactyly | Brachydactyly | Brachydactyly | Brachydactyly | ||
| Meets clinical criteria | YES | YES | YES | YES | YES | YES | YES | NO | YES |
- NA, not available or applicable; NK, not known; NAD, nil abnormalities detected; ND, not done; g, grams; cm, centimetres; SVD, spontaneous vaginal delivery; FTT, failure to thrive; GORD, gastro-oesophageal reflux disease; FRAX, Fragile X; DD, developmental delay; Macrodontia, central incisor width >10 mm in males and >9.7 mm in females; ROP, Retinopathy of Prematurity.
| CASES | % | ANKRD11 mutations-Previously Reported | % | Deletion | % | Overall | % | |
|---|---|---|---|---|---|---|---|---|
| GENDER | ||||||||
| Male | 7/18 | 38.89 | 39/78 | 50.00 | 19/33 | 57.58 | 65/129 | 50.39 |
| Female | 11/18 | 61.11 | 39/78 | 50.00 | 14/33 | 42.42 | 64/129 | 49.61 |
| FACIAL APPEARANCE | ||||||||
| Gestalt | 17/18 | 94.44 | 59/78 | 75.64 | 31/33 | 93.94 | 105/129 | 81.40 |
| Cranial size/ shape anomaly | 15/18 | 83.33 | ||||||
| Brachy/turricephaly | 5/18 | 27.78 | ||||||
| Broad/round/triangular | 13/18 | 72.22 | ||||||
| Hypertelorism/telecanthus | 14/18 | 77.78 | ||||||
| Low hairline | 8/18 | 44.44 | ||||||
| Broad/bushy eyebrows | 8/18 | 44.44 | ||||||
| Synophrys | 7/18 | 38.89 | ||||||
| Strabismus | 3/18 | 16.67 | ||||||
| Ptosis | 3/18 | 16.67 | ||||||
| Any Ear Anomaly | 16/18 | 88.89 | ||||||
| Upturned/anteverted nares | 8/18 | 44.44 | ||||||
| High/prominent bridge | 10/18 | 55.56 | ||||||
| Bulbous tip | 8/18 | 44.44 | ||||||
| Mouth and Palate | ||||||||
| Thick vermillion | 6/18 | 33.33 | ||||||
| Thin vermillion | 8/18 | 44.44 | ||||||
| Long/flat/hypoplastic philtrum | 12/18 | 66.67 | ||||||
| Palatal irregularity | 10/17 | 58.82 | 15/71 | 21.13 | 7/33 | 21.21 | 32/121 | 26.45 |
| DENTAL | ||||||||
| Macrodontia | 11/12 | 91.67 | 67/81 | 82.72 | 17/26 | 65.38 | 95/119 | 79.83 |
| Fused incisors | 2/18 | 11.11 | ||||||
| Oligodontia | 1/18 | 5.56 | ||||||
| Crowding | 5/18 | 27.78 | ||||||
| Premature Decay | 3/18 | 16.6 | ||||||
| Cleft teeth | 0/12 | 0.00 | ||||||
| Other dental, total | 5/18 | 27.78 | ||||||
| HAND ANOMALIES − ALL | 15/18 | 83.33 | 50/71 | 70.42 | 19/29 | 65.52 | 84/118 | 71.19 |
| Brachydactyly | 11/18 | 61.11 | ||||||
| 5th finger clinodactyly | 11/18 | 61.11 | ||||||
| Syndactyly | 5/18 | 27.78 | ||||||
| NEUROLOGICAL | ||||||||
| Delayed fontanelle closure | 2/18 | 11.11 | 7/32 | 21.88 | 9/50 | 18.00 | ||
| Hypotonia | 4/18 | 22.22 | ||||||
| Intellectual disability | 18/18 | 100.00 | 80/82 | 97.56 | 29/30 | 96.67 | 127/130 | 97.69 |
| Seizures | 3/18 | 16.67 | 17/60 | 28.33 | 10/33 | 30.30 | 30/111 | 27.03 |
| Autism Spectrum Disorder [ASD] | 1/18 | 5.56 | 21/97 | 21.65 | 22‘/115 | 19.13 | ||
| ADHD/significant behavioural issues | 10/18 | 50.00 | 50/71 | 70.42 | 17/32 | 53.13 | 77/121 | 63.64 |
| Abnormal MRI/CT brain findings | 5/6 | 83.33 | ||||||
| Any visual abnormality | 10/18 | 55.56 | ||||||
| Hearing loss | 6/18 | 33.33 | 19/71 | 26.76 | 8/32 | 25.00 | 33/121 | 27.27 |
| Congenital heart disease | 4/18 | 22.22 | 14/71 | 19.72 | 8/33 | 24.24 | 26/122 | 21.31 |
| Cryptorchidism | 3/7 | 42.86 | 11/26 | 42.31 | 2/10 | 20.00 | 16/43 | 37.21 |
| Inguinal hernia | 3/18 | 16.67 | 2/51 | 3.92 | 5/69 | 7.25 | ||
| SKELETAL ANOMALY, any | 14/18 | 77.78 | ||||||
| Stature <3rd percentile | 8/18 | 44.44 | 39/82 | 47.56 | 17/32 | 53.13 | 64/132 | 48.48 |
| Delayed bone maturation | 5/10 | 50.00 | 15/36 | 41.67 | 3/16 | 18.75 | 23/62 | 37.10 |
| Accessory ribs | 1/16 | 6.25 | ||||||
| Abnormal vertebra shape | 2/16 | 12.50 | 14/26 [Ockeloen] | 53.85 | 1/14 | 7.14 | 17/56 | 30.36 |
| Osteopenia | 3/16 | 18.75 | ||||||
| Hip anomaly | 2/16 | 12.50 | ||||||
| Kyphosis/scoliosis | 5/18 | 27.78 | ||||||
| Clavicular pseudoarthrosis | 1/16 | 6.25 | ||||||
| Hypoplastic ribs | 1/16 | 6.25 | ||||||
- Bold entries denote more common clinical observations.
| Patient | 1.1 | 1.2 | 1.3 | 2.1 | 3.1 | 3.2 | 3.3 |
|---|---|---|---|---|---|---|---|
| Codon | c.1903_1907 del | c.1903_1907 del | c.1903_1907 del | c.505G>T | c.4406G>A | c.4406G>A | c.4406G>A |
| Genomic coordinates | g.89351043_89351047del | g.89351043_89351047del | g.89351043_89351047del | g.89357129C>A | g.89348544 C>T | g.89348544 C>T | g.89348544 C>T |
| Protein | pLys635GInfs*26 | pLys635GInfs*26 | pLys635GInfs*26 | p.Glu169* | p.Trp1469* | p.Trp1469* | p.Trp1469* |
| Exon | 10 | 10 | 10 | 7 | 10 | 10 | 10 |
| Effect | Frameshift | Frameshift | Frameshift | nonsense | nonsense | nonsense | nonsense |
| Novel/known | known | known | known | Novel | Novel, Inherited | Novel, Inherited | Novel |
| Sequencing test | Single gene | Single gene | Single gene | Single gene | Single gene | Single gene | Single gene |
| Patient | 4.1 | 5.1 | 7.1 | 8.1 | 9.1 | 11.1 | 12.1 | 12.2 | 13.1 |
|---|---|---|---|---|---|---|---|---|---|
| Codon | c.1903_1907del | c.1173C > G | c.7471A>C | c.6409_6410del | c.3224_3227delCTTT | c.7216C > T | c.3442G > A | c.3442G > A | c.6472G > T |
| Genomic Coordinates | g.89351043_89351047del | g.89351777G > A | g.89341601T > G | g.89346540_89346541del | g.89349723_89349726delAAAG | g.89346734G > A | g.89349508C > T | g.89349508C > T | g.89346478C > A |
| Protein | p.lys635GLnfs*26 | p.Tyr391* | ? | p.Ser2137Profs*9 | p.Glu1075Glyfs*242 | p.Gln2406* | p.Gly1148Ser | p.Gly1148Ser | p.Glu2158* |
| Exon | 10 | 10 | Splice site acceptor exon 11 | 10 | 10 | 10 | 10 | 10 | 10 |
| Effect | Frameshift | nonsense | Splice site | Frameshift | Frameshift | Nonsense | Missense | Missense | Nonsense |
| known | Novel, de novo | Novel, de novo | Novel, de novo | Novel, de novo | Novel, de novo | Novel, inherited | Novel | Novel, de novo | |
| Sequencing test | Intellectual disability panel | Single gene | Single gene | Single gene | WES | WGS | Intellectual Disability panel | Segregation | WES |
- Reported with respect to transcript NM001256182.1 and genome assembly GRCh37/HG19.
- WES, whole exome sequencing; WGS, whole genome sequencing.
- Patients 6.1 and 10.1 have not yet undergone genetic testing.




4 RESULTS AND DISCUSSION
4.1 Clinical and radiological observations
Of our 18 patients, all but one met minimum clinical diagnostic criteria albeit with significant inter- and intra-familial variability.
4.1.1 Phenotype
Seventeen patients had the facial appearance characteristic of KBG syndrome. This concurred with previous reports (Goldenberg et al., 2016 ; Low et al., 2016 ) which showed characteristic facies in 75% of mutation and 89% of deletion cases, including those identified through the DDD study. The commonest dysmorphic features in our patients included ear abnormalities in 16/18 [89%], and the presence of cranial shape anomalies in 15/18 patients [83%]. A prominent nasal bridge was present in 10/18 [56%] patients, while hypoplastic alae nasi were present in 4/18 [22%]. Abnormal philtrum length and morphology was detected in 12/18 patients [66.7%] while 14/18 [77.8%] had hypertelorism.
4.1.2 Ear, nose, and throat aspects
Hearing loss was reported in 6/18 [33%] compared with 27% in previous cases. Interestingly, a case of severe hearing loss including a sensorineural component was reported in a patient with the recurrent 1903_1907 deletion which was previously associated with a milder hearing loss phenotype (Low et al., 2016 ). Patient AU3.2 had unilateral hearing loss since birth, not otherwise specified. The remaining patients had a purely conductive pattern of hearing loss (Table 1 ).
Ten patients of 17 [58.9%] had an abnormality of the palate, which was higher than in previous reports of both mutation and deletion patients [21.1%]. Unrelated patients AU2.1 and AU5.1 had a bifid uvula, and patients 3.1 and 3.3 had an incomplete palate diagnosed endoscopically. The remaining anomaly was presence of a high arched palate.
4.1.3 Dental aspects
Dental findings in this cohort were similar to those previously described and included macrodontia* in 11/12 patients [91.7%], excluding those patients with primary dentition. Other dental abnormalities included premature tooth loss in patients AU1.1, AU3.2, and AU3.3 [16.6%], and crowding in 5/18 [27.8%] patients. Patients AU1.3 and AU8.1 had fusion of the permanent maxillary central incisors.
4.1.4 Hand anomalies
Hand anomalies were common in 15/18 [83%], higher than in previous reports [70% of mutation and 65.5% of deletion cases]. Brachydactyly and 5th finger clinodactyly [11/18 cases each] were the commonest; while cutaneous syndactyly was identified in 5/18 cases [27.78%].
4.1.5 Neurological and psychiatric features
Intellectual disability ranging from mild to severe was present in all 18 of our patients, compared with previous reports [97.7%]. Nine patients, including half-brothers AU1.1 and 1.2, had significant language delay out of proportion to performance on standardized cognitive testing. Only one of these patients met DSM-V criteria for autism spectrum disorder. This concurs with the findings of Low et al. (2016 ), which also reported disproportionate language deficits. Only three of our cohort had seizures diagnosed at the time of writing (16.7%), compared with 28.3% of patients with ANRKD11 mutations reported previously, and 30.3% of patients with a contiguous gene deletion at the 16q23 region. The previously identified pattern of seizures improving with age (Ockeloen et al., 2015 ) was observed patient AU5.1, who was seizure free by the age of 6. Six patients in this series underwent MRI brain scanning. Patient AU5.1 had an MRI at the age of 4, showing small arachnoid and pineal cysts, while patient 4.1 had a low lying conus medullaris, partial agenesis of the corpus callosum, and choroid plexus cysts. Patient AU1.2 had small optic nerves and volume loss with a developmental venous anomaly in the frontal lobe. Patient AU11.1 had an MRI showing a dilated left sided endolymphatic sac and arachnoid cysts overlying the cerebellar hemispheres. MRI for patient AU13.1 was markedly abnormal and showed bilateral T2 hyperintensities, caudate swelling, and high signal and calcific foci in the periventricular white matter and right thalamus. Arachnoid cysts were also present. No environmental cause was found to explain these findings and whole exome sequencing did not reveal an alternative genetic explanation. Results from the MRI brain of patient 12.1 were unremarkable.
Two patients in this series had the newly recognized feature of a large anterior fontanelle and delayed closure.
Significant behavioral and psychiatric abnormalities are increasingly recognised in this syndrome (Ockeloen et al., 2015 ) and were evident in 50% of our patients. Physical aggression was reported in three of our patients [16.7%] along with reports of temper tantrums and inflexible personality traits in patients 3.1 and 8.1, and a clinical diagnosis of Bipolar Affective Disorder in patient 1.3. There did not appear to be a correlation between behavior with the degree of intellectual disability, and further research is needed to delineate the behavioral profiles manifest in KBG syndrome. Previous reports suggested major behavioral problems in 70% of those with a mutation and 53% with a deletion of ANKRD11. Visual anomalies were present in 10/18 patients [56%], with refractive errors or astigmatism, 2 of whom had concurrent strabismus. Low et al. (2016 ) reported a similar frequency of refractive errors in their study.
4.1.6 Cardiac features
Congenital heart disease is a feature of mutation positive KBG syndrome, present in 4/18 [22%] unrelated patients as reported previously. Patient AU3.3 had an atrial septal defect and ventriculoseptal defect [VSD] repairs in childhood while Patient 1.2 had a VSD and patent ductus arteriosus on an echocardiogram in infancy. Patient 6.1 had a primum atrial septal defect and a cleft mitral valve while patient 12.1 had a persistent patent ductus arteriosus.
4.1.7 Genitourinary features
Cryptorchidism was identified previously in 16/17 [97%] of patients s with a mutation and in 2/10 with a deletion of ANKRD11. 3/7 of our male patients required operative management to correct cryptorchidism in childhood, reiterating that this is a common phenotypic feature. One patient had precocious puberty, a finding reported in 5/39 patients in a recent review (Goldenberg et al., 2016 ).
4.1.8 Skeletal aspects
The presence of skeletal features including stature <3rd centile, delayed bone maturation, and costovertebral anomalies form three of the major diagnostic criteria for KBG syndrome (Skjei et al., 2007 ). Only 8/18 of our patients [44%] had postnatal short stature <3rd centile, with an additional patient on the tenth centile, and two patients had intrauterine growth restriction [IUGR]. The remaining patients ranged between the 50th and 75th centile, indicating that postnatal short stature may not be as common in Australian patients with this syndrome.
The costovertebral anomalies we observed were variable and generally mild. Four of 16 patients had a normal survey other than the presence of short metacarpals and metatarsals, while an additional three patients had only mild scoliosis or lordosis. Patient AU4.1 had bilateral coxa valga, while patient AU7.1 had narrow spaces in the thoracic spine, and symphalangism in toes 3–5 along with congenital bilateral hip dislocation. Patient AU6.1 had marked pectus excavatum and an exaggerated lumbar lordosis. The rates of vertebral abnormalities were higher in the patients published by Ockeloen and Sirmaci [14/26, 53.85%], and hip abnormalities were more common [16%]. Delayed bone maturation was evident in 5/10 paediatric patients and osteopenia was also identified in three children. Our findings suggest wide variability in the skeletal manifestations of this condition, and that costovertebral abnormalities may be less prevalent than previously thought.
4.1.9 New features identified
Additional findings in our patients included aberrant abdominal vessels and malrotation of the abdominal viscera in patient AU4.1, who required a PEG tube for feeding. She also had ectopic kidneys identified on ultrasonography. A malrotated bowel requiring corrective surgery at 10 weeks was also identified in patient AU12.1. Novel skeletal findings included the presence of clavicular pseudoarthrosis in a patient and the presence of bilateral cervical ribs. Osteopenia was also identified in three paediatric patients, including a male patient 1.2 at the age of 5 years, and two female patients 2.1 and 18.1 at the ages of 13 and 6 years, respectively. The unusual MRI appearance in patient AU13.1 of widespread calcific changes and basal ganglia abnormalities did not have an identified secondary cause. Another unique finding was of microphthalmia in the same patient and corneal clouding described in an infant. Two patients were identified as having growth restriction in utero.
To our knowledge, none of these findings have been reported previously in patients with an ANKRD11 mutation or deletion.
There was no consistent phenotype-genotype correlation.
4.1.10 Intrafamilial observation
Family 1 included two half-brothers who had similar facial features brachydactyly and abnormalities of the palate. Both had significant learning difficulty, with expressive and receptive language impairment and behavioral problems. The older boy, assessed at age 7, had a history of cryptorchidism and bilateral hearing loss as well as macrodontia. The younger half-brother was a dichorionic diamniotic twin born prematurely at 27 weeks. At the age of 4 years, he had growth delay, osteopenia, and his echocardiogram showed a patent ductus arteriosus and ventricular septal defect. His clinical picture was confounded by complications of prematurity including failure to thrive and grade IV retinopathy. Their mother had a similar facial appearance. She was shorter than her sisters. She had a mild scoliosis and fusion of the central incisors with macrodontia. She had a history of learning difficulty and bipolar affective disorder. The recurrent point mutation c.1903_1907del [g.89351043_89351047del pLys635GInfs*26] segregated in these three family members.
Family 2 had three generations of affected individuals sharing similar dysmorphic features, nasal speech with palate incompetence, brachydactyly, and intellectual disability. Individual 3.1, examined at age 13, had macrodontia, a unilateral strabismus along with behavioral issues including aggression and inflexibility. Her height was on the 75th centile, while her mother [3.2] and grandmother [3.3] had heights on the 3rd centile. A 3.2 had the additional features of congenital right sided hearing loss and a hypoplastic 12th rib, while individual 3.3 had a childhood history of repaired atrial and ventricular septal defects. 3.2 and 3.3 had premature decay of permanent dentition. A novel nonsense mutation [g.89348544 C>T p.Try1469*] segregated with KBG syndrome in this family.
Family 12 came to attention with the clinical diagnosis of KBG syndrome in individual 12.1, who had conductive hearing loss, strabismus, intellectual disability with language delay, and dysmorphism including brachydactyly, syndactyly, a webbed neck, and a broad forehead. Other features included a malrotated bowel at 10 weeks of age, along with delayed bone age and a patent ductus arteriosus. A novel missense mutation was detected in exon 10 of ANKRD11 [g.89349508C>T; p.Gly1148Ser], and identified in her father [12.2] on segregation studies. A 12.2 did not meet diagnostic criteria although he had some consistent facial features and a history of learning difficulty. He had no apparent skeletal and cardiac anomalies and his height was at 90th centile.
4.1.11 Revised diagnostic criteria
Recently proposed revisions to the Skjei criteria are published previously (Low et al., 2016 ; Skjei et al., 2007 ).
We agree with the authors that stature below the 10th centile is more appropriate than stature below the 3rd centile as a diagnostic criterion. However, 10/18 of our patients [66%] had a stature on or above the 10th centile, with two patients on the 75–90th centile. This suggests that short stature is not as prevalent in Australian patients with KBG syndrome. We also agree with the removal of costovertebral anomalies from the diagnostic criteria as we found these to be variable and mild, as reported by others (Goldenberg et al., 2016 ; Low et al., 2016 ). We agree with the inclusion of palate abnormalities as a diagnostic aid, as these were found in 58% of our patients and 26% overall. Most of our patients had typical hand anomalies [83.3%] and neurological features including learning disability [100%] and we recommend that these remain as principle features in standard diagnostic criteria.
Our patients deviated from reported data in the low incidence of seizures [16.7%], although EEGs were not performed in most. The low frequency of autism spectrum disorder reflects the findings in other report (Goldenberg et al., 2016 ) but it represents a departure from the previous estimates of 25% (Low et al., 2016 ) and 47% (Ockeloen et al., 2015 ).
4.2 Molecular results (Table 3)
Mutations in the ANKRD11 gene were reported with respect to genome assembly GRCh37/Hg19 and transcript NM001256182.1. The UCSC browser was used in conjunction with the Alamut database. SIFT, Polyphen, and AlignGVGD were used for in silico prediction of pathogenicity for the novel missense mutation reported in patients AU12.1 and 12.2. Five nonsense, four frameshift mutations, one splice site mutation, and one missense mutation were identified. Nine novel mutations were identified. The p.Glu169* variant in AU2.1 truncates the protein from the first (amino acids 167-196) of the four ANK domains of ANKRD11. It is the closest variant to the N-terminal of the protein reported to date. It has been shown that ANKRD11 protein homodimerizes through the ANK domains located at the N-terminal. Since, all other mutations reported to date left the N-terminal of the protein intact, it has been hypothesized that the truncated ANKRD11 may bind the normal ANKRD11 via its N-terminal, potentially impairing the function of the normal copy. The presence of typical manifestations of KBG syndrome in our patient 2.1 in whom complete nonsense mediated mRNA decay was expected supported ANKRD11 haploinsufficiency in causation of KBG syndrome. This is an interesting finding that is against the hypothesis of dominant negative mechanism acting in the pathogenesis of KBG syndrome.
There was no observed correlation between phenotype and the site or domain at which protein truncation was predicted (Figure 4 ). Features and severity did not appear to correlate with mutations in the de novo form although the father AU12.2 did not meet clinical criteria and had a pathogenic mutation detected on segregation studies, suggesting a milder effect conferred by this missense mutation. The findings confirm specificity of ANKRD11 mutations in KBG syndrome as well as allelic heterogeneity.
5 CONCLUSION
Our 18 Australian patients with KBG syndrome including 16 with a mutation in ANKRD11 highlights several key aspects of KBG syndrome, including the ongoing clinical utility of the diagnostic criteria with potential revisions accounting for normal stature and infrequency of costovertebral anomalies. Diagnosis may be delayed in the proband and examination of parental dentition may provide an essential clue.
Language delay is severe and out of proportion to deficits in other domains and the behavioral features do not appear to correlate with the degree of intellectual disability. We recommend echocardiogram, renal ultrasonography, ophthalmic, and hearing assessments and good dental care along with formal developmental assessments and appropriate early intervention. We also recommend bone density measurement because of increased frequency of osteopenia (16%). There is significant allelic heterogeneity in this condition. The results confirm the specificity of ANKRD11 mutations in KBG and provide further evidence for this transcription repressor in neural, cardiac, and skeletal development. Further mutation testing of clinically affected individuals will assist in broadening our understanding of the phenotype and genotype-phenotype correlations, in particular the skeletal and dental manifestations of this syndrome as well as the neurobehavioral phenotype.
Disclosures: Nothing to disclose. Ethics committee approval was not required as the manuscript is a retrospective case series. Documented permission from patients regarding the use of images is attached. All contributors have read and approved submission to the journal.