Corner fracture type spondylometaphyseal dysplasia: Overlap with type II collagenopathies
Abstract
Spondylometaphyseal dysplasia (SMD) corner fracture type (also known as SMD “Sutcliffe” type, MIM 184255) is a rare skeletal dysplasia that presents with mild to moderate short stature, developmental coxa vara, mild platyspondyly, corner fracture-like lesions, and metaphyseal abnormalities with sparing of the epiphyses. The molecular basis for this disorder has yet to be clarified. We describe two patients with SMD corner fracture type and heterozygous pathogenic variants in COL2A1. These two cases together with a third case of SMD corner fracture type with a heterozygous COL2A1 pathogenic variant previously described suggest that this disorder overlaps with type II collagenopathies. The finding of one of the pathogenic variants in a previously reported case of spondyloepimetaphyseal dysplasia (SEMD) Strudwick type and the significant clinical similarity suggest an overlap between SMD corner fracture and SEMD Strudwick types. © 2016 Wiley Periodicals, Inc.
INTRODUCTION
Spondylometaphyseal dysplasias (SMD) are a heterogeneous group of disorders characterized by vertebral and metaphyseal abnormalities. According to the last Nosology of Constitutional Bone Disorders from 2015 [Bonafe et al., 2015], this group includes eight disorders (including SMD Sedaghatian-like type) classified according to their severity, distinctive skeletal abnormalities (e.g., enchondroma-like lesions in spondylo-enchondrodysplasia), the presence of extra-skeletal manifestations or according to the associated gene, if known. The molecular basis for most of these disorders is known, but the causative gene for some has not been identified yet.
SMD corner fracture type (also referred to as “Sutcliffe” type by Kozlowski et al., 1992) is a rare autosomal dominant skeletal dysplasia characterized by a triad of mild vertebral abnormalities (platyspondyly), developmental coxa vara, and metaphyseal irregularity with corner fracture-like lesions that can be seen through early development and tend to disappear with skeletal maturation [Langer et al., 1990]. Radiologic findings of corner fractures during the first 1–2 years of life are highly concerning for non-accidental trauma [Leaman et al., 2016]. Yet, corner fracture-like lesions can also be detected in the setting of certain skeletal dysplasias, and although considered characteristic for SMD corner fracture type, these radiological findings have been described in other disorders such as metaphyseal chondrodysplasia and spondyloepimetaphyseal dysplasia (SEMD) Strudwick type [Langer et al., 1990;Sutton et al., 2005]. Although the molecular basis for SMD corner fracture type is not yet known, one case report of SMD corner fracture type due to a heterozygous COL2A1 pathogenic variant has been described in the literature [Walter et al., 2007].
Here, we describe two additional cases of SMD corner fracture type with heterozygous pathogenic variants in COL2A1. These cases suggest that at least some cases of SMD corner fracture type are part of the type II collagenopathy spectrum of disorders.
MATERIALS AND METHODS
Human Subjects
Patient 1 was enrolled in a research protocol approved by the Institutional Review Board (IRB) of Baylor College of Medicine. Informed consent for whole exome sequencing (WES) and publication of medical information was obtained. WES for patient 2 was done on a clinical basis. Samples from parents of patient 2 were not available for testing.
Whole Exome and Sanger Sequencing
For patient 1, genomic DNA was isolated from peripheral blood monocytes and WES was done as previously described [Yang et al., 2013]. Oligonucleotide primers (forward—ACACCTCGCCATCCTCGT; reverse—CAAAGCTTGTTATCGATATGCTCA) were designed to amplify exon 17 of COL2A1 (ENST00000380518) in the patient and in his parents using polymerase chain reaction (PCR). Exon 17 was amplified using 2 ng/uL of DNA with GoTaq polymerase (Promega, Madison, WI) and was submitted for Sanger sequencing at Beckman Coulter Genomics (Danvers, MA). For patient 2, WES on genomic DNA from the affected individual and variant filtering was performed as previously described [van Karnebeek et al., 2016]. For PCR amplification, oligonucleotide primers (forward—TAAGCTGCCTGCCCTTAGTGTG; reverse—ATGCTCTGTGAGCTCAGAAGCC) were designed to amplify exon 42 of COL2A1 (ENST00000380518) in the patient.
CLINICAL REPORT
Patient 1
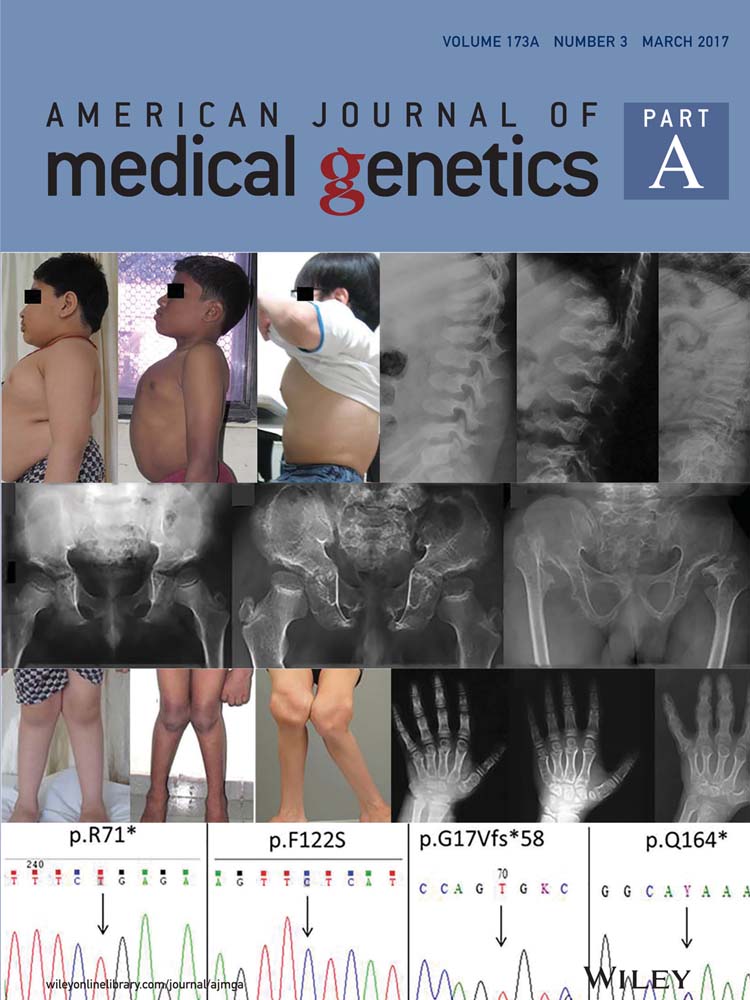
Patient 1 was born full term by spontaneous vaginal delivery. Prenatal history was remarkable for shortening of the long bones on ultrasound performed at 35 weeks of gestation. Postnatal genetic testing for achondroplasia was negative. He was first seen in the Texas Children's Hospital Skeletal Dysplasia Program at the age of 22 months. At that time, his development was reported to be age appropriate. On physical exam, his height was 75.7 cm (Z-score of −3), and his weight was at the 25th–50th centile (12 kg). Head circumference was at the 95th centile (51.2 cm). He had accentuated lumbar lordosis. Lower extremities were markedly short with genu vara bilaterally. Fourth and fifth digit clinodactyly was noted in the hands bilaterally. Radiographs (Fig. 1a–d) revealed findings of SMD. Skeletal survey from the first year of life was notable for platyspondyly as well as significant metaphyseal flaring and corner fracture-like lesions at many of the long bones. The epiphyses appeared spared. Given the findings of short stature, platyspondyly, metaphyseal irregularity, and corner fracture-like lesions, a diagnosis of SMD corner fracture type was given. No coxa vara deformity was present. Surgery was later required for severe, progressive genu valgum. He had no history of joint hypermobility or dislocation, cleft palate, myopia, or hearing deficit. His family history was positive for a father with unilateral slipped-capital femoral epiphysis at the age of 15 years.

Patient 2
Patient 2 was the first boy of healthy non-consanguineous Chinese parents. He was born at term with a birth weight of 2.65 kg (5th centile) and length of 49 cm (25th centile). The mother reported that his short stature was evident from the age of 4 years, but his first formal evaluation was at the age of 11 years. At 11 years, his height was 118.9 cm (Z score of −3.7), and he had a disproportionately short trunk. He had a mild pectus carinatum and scoliosis. He walked with a limp, and radiographs (Fig. 1e–h) revealed significant dysplasia of the femoral neck with corner fracture-like lesions. The epiphyses appeared spared. Given the findings of short stature, platyspondyly, metaphyseal irregularity, right coxa vara, and corner fracture-like lesions, a diagnosis of SMD corner fracture type was given. At age 12 years, he underwent an osteotomy valgisation of the right femur. He had no history of cleft palate, myopia, or hearing deficit. His parents’ heights were 152 cm (mother) and 170 cm (father).
RESULTS
WES for patient 1 revealed a heterozygous missense variant in COL2A1. Sanger sequencing in the proband and his parents revealed that this variant is de novo. This variant causes the replacement of the amino acid glycine in position 345 with aspartic acid (c.1034 G>A, p.Gly345Asp) and was reported once previously in a cohort of 71 patients with skeletal dysplasia (no detailed phenotype was published, Barat-Houari et al., 2016a). WES for patient 2 revealed a heterozygous missense variant in COL2A1. This variant causes the replacement of the amino acid glycine in position 945 with serine (c.2833G>A, p.Gly945Ser) and was reported previously in a Dutch family diagnosed with mild spondyloepiphyseal dysplasia congenita (SEDC) [Terhal et al., 2015]. Patients in this family were reported to have mildly short to normal stature, scoliosis, coxa vara and minimal bilateral cataract.
Both variants do not appear in NCBI dbSNP or in ExAc database [http://exac.broadinstitute.org, Lek et al., 2016]. Both changes are predicted to be pathogenic as they are predicted to disrupt COL2A1 homotrimer assembly and stability [Mouw et al., 2014], and both meet ACMG guidelines for pathogenicity [Richards et al., 2015]. PolyPhen2 [http://genetics.bwh.harvard .edu/pph2] and SIFT [http://sift.jcvi.org/], two software programs that are used to predict the functional effect of an amino acid substitution, also predict that both variants are pathogenic.
DISCUSSION
SMD is a heterogeneous group of rare skeletal dysplasias characterized by dysplastic development of the spine and generalized metaphyseal modifications of the tubular bones. The disorders in this group are diverse in their clinical presentation, and the molecular basis for six of the disorders in the SMD group is known (Table I). SMD Algerian type and dysspondylo-enchondromatosis (DSC) were previously considered as distinct entities in the SMD group of disorders. However, identifying patients with COL2A1 pathogenic variants and significant clinical overlap with SEMD Strudwick type (type II collagenopathy) changed their classification, and they are currently part of the type II collagenopathy spectrum [Bonafe et al., 2015].
| Kozlowski | Sedaghatian | Severe SMD (Sedaghatian-like) | ODCD | SMD-CRD | SMD with retinal degeneration, axial type | SPENCD | Corner fracture | SEMD Strudwick type | ||
|---|---|---|---|---|---|---|---|---|---|---|
| OMIM # | 184252 | 250220 | None | 184260 | 608940 | 602271 | 271550 | 184255 | 184250 | |
| 2015 Nosology group (#) | TRPV4 (8) | Severe SDD (14) | Severe SDD (14) | SMD (12) | SMD (12) | SMD (12) | SMD (12) | SMD (12) | Type 2 collagen (2) | |
| Inheritance | AD | AR | AR | AR | AR | AR | AR | AD | AD | |
| Short stature | + | + | + | + | + | + | + | + | + | |
| Short trunk | + | + | + | + | + | + | ||||
| Short limbs | + (R) | + (R) | + (M) | + (R) | + (R) | + (R) | + | |||
| Metaphyseal changes | Irregular metaphysis | + | + | + | + | + | + (Proximal femur) | + | + | + |
| Corner fracture type lesions | + | + [Langer et al., 1990] | ||||||||
| Epiphyseal involvement | + | + | + | + | ||||||
| Other bone abns. | Enchondroma-like lesions | + | ||||||||
| Vertebral abns. | Scoliosis | + | + | + | + | + | ||||
| Platyspondyly | + | + | + | + | + | + | + | + | + | |
| Odontoid hypoplasia | + | + | + | |||||||
| Hip abns. | Coxa vara | + | + | + | + | + | ||||
| Iliac bone abnormality | + | + | + | + | + | + | ||||
| Chest | Pectus carinatum | + | + | + | + | |||||
| Small chest | + | + | ||||||||
| Rib abnormalities | + | + | + | + | + | |||||
| Leg abns | + (Genu vara/recurvata) | + (Bowing) | + (Genu valgus) | |||||||
| Hands | Irregular carpal/metacarpal bones | + | + | + | ||||||
| Brachydactyly | + | + | + | + (Short distal phalanges) | + | |||||
| Feet | Short and thick feet/Clubfeet | + | + | |||||||
| Joints | Hyper extensible joints | + | ||||||||
| Skull | Skull abnormalities | + | + | |||||||
| Face | Facial dysmorphism | + | + | + | + (Cleft palate) | |||||
| Dental | Dentinogenesis imperfecta | + | ||||||||
| Eyes | Vision impairment | + | + | + (Myopia) | ||||||
| Hearing | Hearing impairment | + | ||||||||
| Skin | + (Redundant skin) | +(Pigmentation abns) | ||||||||
| Systemic involvement | Respiratory problems | + (Pulmonary hemorrhage) | + | + | + (Recurrent pneumonia) | + (Neonatal period, interstitial fibrosis) | ||||
| Hypotonia | + | + | ||||||||
| Cardiac | + (Conduction defects) | |||||||||
| CNS | + | + (Seizures) | + | |||||||
| Immunodeficiency with autoimmunity | + (Neutropenia) | + | ||||||||
| Intellectual disability | + | |||||||||
| Lethality | + | |||||||||
| Molecular basis | TRPV4 | GPX4 | SBDS | Unknown | PCYT1A | C21orf2 | ACP5 | COL2A1 | COL2A1 | |
| Reference | Krakow et al. [2009] | Smith et al. [2014] | Nishimura et al. [2007] | Hoover-Fong et al. [2014] | Wang et al. [2016] | Lausch et al. [2011]; Briggs et al. [2011] | Walter et al. [2007] | Tiller et al. [1995] |
- SEMD included (gray column) for comparison purposes.
- SMD, spondylometaphyseal dysplasia; SDD, spondylodysplastic dysplasias; ODCD, odontochondrodysplasia; SPENCD, spondyloenchondrodysplasia; SEMD, spondyloepimetaphyseal dysplasia; M, mesomelic; R, rhizomelic; SMD-CRD, SMD with cone-rod dystrophy; AD, autosomal dominant; AR, autosomal recessive; abns, abnormalities; CNS, central nervous system.
Walter et al. [2007] previously described a 7-year-old female with SMD corner fracture type who was found to have a heterozygous missense pathogenic variant in COL2A1 (p.Gly181Arg, amino acid numbering as was published, starts with the first glycine of the triple helix). As in our two patients, she presented with metaphyseal irregularities, vertebral abnormalities, and corner fracture-like lesions (Table SI, supplementary data). All three patients have heterozygous missense pathogenic variants in COL2A1 that result in substitution of a glycine residue. The pathogenicity of these variants is almost certain as glycine plays a conserved and crucial role in the helical Gly-X-Y repetitive sequence of the collagen helix.
Pathogenic variants in type II collagen cause a wide spectrum of skeletal dysplasias that was reviewed previously elsewhere [Barat-Houari et al., 2016b]. This wide spectrum of overlapping clinical presentations associated with pathogenic variants in type II collagen has only limited phenotype–genotype correlation. The rarity of the disorders and the difficulty in diagnosis by non skeletal dysplasia experts [Zhang et al., 2015] make it difficult to determine the phenotypic spectrum of specific skeletal dysplasias. The diagnostic challenge with SMD corner fracture type is even greater as corner fracture-like lesions tend to disappear with skeletal maturation and the severity of the metaphyseal involvement tends to decrease with age. Although developmental coxa vara is considered to be one of the central features in SMD corner fracture type, our first patient did not have obvious femoral neck abnormality. Langer et al. [1990] described a series of patients with SMD corner fracture type. While most of the patients had bilateral developmental coxa vara, one patient had only unilateral findings, raising the possibility of wider clinical spectrum.
Barat-Houari et al. [2016a] published a cohort of 121 patients with skeletal dysplasia. Seventy-one of these patients were found to have a COL2A1 pathogenic variant and eight of these patients (11%) had the diagnosis of SEMD Strudwick type. Interestingly, one of the eight patients was reported to have the same pathogenic variant as our first patient (p.Gly345Asp). SMD corner fracture type shares significant clinical features with SEMD Strudwick type including metaphyseal abnormalities, coxa vara, platyspondyly, and odontoid hypoplasia (Table I). Moreover, corner fracture-like lesions have also been described in SEMD Strudwick type. These findings suggest clinical overlap between SMD corner fracture type and SEMD Strudwick type.
The finding of three patients with clinical diagnosis of SMD corner fracture type and pathogenic variants in COL2A1, two of which had been previously described in patients with type II collagenopathies, may suggest that this type of SMD is a heterogeneous disorder with a subset of patients showing overlap with type II collagenopathies. Thus, COL2A1 molecular testing should be considered in patients with the finding of corner fracture-like lesions in the settings of SMD.
ACKNOWLEDGMENTS
We thank the patients and their families. We thank Dr. Magdalena Walkiewicz for her technical assistance. This work was supported by the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH U54HG003273 (R.A.G.), NIH U54 U54HG006542 (R.A.G.), NIH T32 GM07526 (M.J.), NIH K08DK106453 (L.C.B), and NICHD P01 HD070394 (B.L).