Further evidence for GRIN2B mutation as the cause of severe epileptic encephalopathy
Abstract
Epileptic encephalopathies (EE) include a range of severe epilepsies in which intractable seizures or severe sub-clinical epileptiform activity are accompanied by impairment of motor and cognitive functions. Mutations in several genes including ion channels and other genes whose function is not completely understood have been associated to some EE. In this report, we provide a detailed clinical description of a sporadic male patient with early-onset epilepsy and epileptic encephalopathy in whom we performed complete exome sequencing (WES) and identified a GRIN2B mutation. The GRIN2B splicing mutation in intron 10 (c.2011-1G>A) was revealed in a WES study. The result was confirmed by Sanger sequencing. No mutation was found in both parents. Our finding confirms that early-onset EE may be caused not only by gain-of-function variants but also by splice site mutations—in particular those affecting the splice acceptor site of the 10th intron of the GRIN2B gene. © 2016 Wiley Periodicals, Inc.
INTRODUCTION
Epileptic encephalopathies (EE) constitute a group of disorders in which severe epilepsy itself is considered as a contributing factor to severe cognitive impairment. The cause of intellectual disability associated with severe epilepsy is often unknown [Berg et al., 2010].
Genome-wide studies using next generation sequencing (NGS) identified an increasing number of causes of developmental and seizure disorders and have revealed significant associations and rare SNV (single nucleotide variants) explaining many cases of epilepsy [de Ligt et al., 2012; Epi4K Consortium et al., 2013; EuroEPINOMICS-RES Consortium et al., 2014, Hamdan et al., 2014]. Many of these mutations seem to converge on specific biological pathways which may be useful for drug development and the personalization of treatment [EuroEPINOMICS-RES Consortium et al., 2014; Pierson et al., 2014]. Up to now, it has been proven that early-onset EE can be associated with mutations (also de novo) in genes coding for proteins involved in neuronal functions and development, synapses and neurotransmitters functions, and, importantly, numerous ion channels [Oliver et al., 2014]. One of the ion channel genes is GRIN2B, which encodes the NMDA (N-methyl-D-aspartate) receptor subunit NR2B, which was found to be disrupted by chromosome translocation breakpoints in individuals with intellectual disability and/or epilepsy [Endele et al., 2010]. NMDA receptors mediate excitatory neurotransmission in the mammalian brain [Cull-Candy et al., 2001; Lau and Zukin, 2007].
By applying NGS-based complete exome analysis (WES), we demonstrated a de novo GRIN2B mutation in a patient with early-onset epileptic encephalopathy.
METHODS
Patient
We investigated a sporadic case of a 6-year-old boy who presented early-onset epileptic encephalopathy without congenital defects of internal organs and without specific facial dysmorphism, including microcephaly. Additionally, we studied the healthy parents of the proband. Venous blood samples were collected from all the above mentioned participants of the study. Written informed consent was obtained prior to genetic testing from the parents (the legal guardians of the proband).
Genetic Testing
The following tests were performed in the proband before WES and yielded normal results: karyotype (normal male), a methylation test, and MLPA assay with telomeric probes. Array CGH revealed microdeletion at the 4q28.1 chromosome containing 450 kb; the same mircrodeletion was observed in the healthy father and sisters of the proband. The deleted region observed in aCGH was not pathogenic according to literature.
NGS analysis was performed using a TruSeq Exome Enrichment Kit according to the manufacturer's instructions (Illumina). The sample was run on 1/4 of a lane on a HiSeq 1500 using 2 × 100 bp paired-end reads. Bioinformatics analysis was performed as previously described [Ploski et al., 2014]. Briefly, after initial processing with CASAVA, the sequencing reads were aligned to the hg19 reference genome with Burrows–Wheeler Alignment Tool and further processed by a Genome Analysis Toolkit [McKenna et al., 2010]. Base quality score recalibration, indel realignment, duplicate removal, and the SNP/INDEL calling were conducted as described [DePristo et al., 2011]. The detected variants were annotated using Annovar and converted to the MS Access format for final manual analyses. Alignments were viewed with an Integrative Genomics Viewer [Wang et al., 2010; Robinson et al., 2011]. Splice site prediction was performed using NetGene2 [Brunak et al., 1991].
Sanger sequencing was performed using a BigDye Terminators kit v 3.1 (Life Technologies) with the following primers: forward 5′GGCATGCATTTCTGCATAGTT3′, Reverse: 5′GCAATTGCTGCTCTTGAGTG3′). Paternity verification was performed using a PowerPlex 16 kit (Promega) for co-amplification of 16 hyper-variable short tandem repat (STR) loci. Capillary electrophoresis, both for Sanger sequencing and paternity verification, was performed using ABI 3700 (Life Technologies).
RESULTS
Clinical Report
The proband was the first male child of a healthy non-consanguineous couple. The proband's family history was non-contributory, with his three younger sisters being healthy. The boy was born at 38 weeks of gestation by natural delivery after an uneventful pregnancy with the following birth parameters: weight 3,400 g, length 53 cm, OFC 31 cm, Apgar score 10 points. No congenital defects were noted after birth. Developmental milestones were severely delayed from newborn period. The child is still not walking or speaking. Epileptic seizures have been observed from the 6 month of age. Seizures are focal and during paroxysmal episodes the boy presents unilateral facial clonic seizures and muscles’ rigidity, however he remains responsive to his parents. Seizures are frequent being observed up to several times per day. The child requires complex multidrug therapy. Treatment with valproic acid and levetiracetam led to no clinical improvement. Replacement of valproic acid with lamotrigine improved neither the seizures (reduction in seizure frequency) nor the EEG (electroencephalography) pattern reaching a plateau in terms of severity of the clinical condition and then remaining stable.
Brain and spinal cord NMR (nuclear magnetic resonance) studies, as well as electromyography, showed no disturbances. MS/MS (tandem mass spectrometry) and GC/MS (gas chromatography—mass spectrometry) assays revealed no abnormalities; lactic acid, CPK, and NH3 levels were in the normal range.
The boy had never a video EEG. An interictal EEG recording during sleep revealed numerous and periodically almost continuous abnormalities in the fronto-temporo-parietal region and temporo-parieto-occipital regions of both hemispheres. There was also generalized paroxysmal activity—generalized sharp waves, sharp wave/slow wave complexes, spike-wave discharges, and theta waves with amplitudes up to 400 uV. No effect of photostimulation was observed. The EEG was performed several times (at the age of 6 months and 2 and 4 years) and always showed abnormal results which were not changed overtime but without any specific pattern that could suggest a particular type of epilepsy.
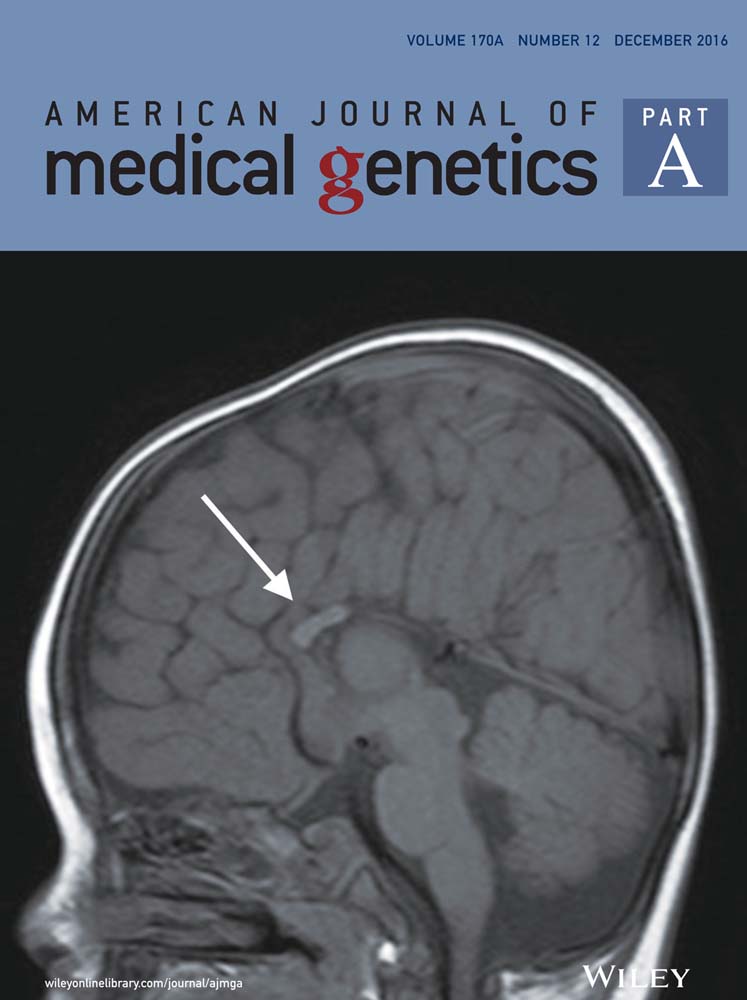
At the last follow-up, at the age of 6 years, the proband's weight was 17 kg (10th percentile), length 115 cm (25th percentile), and head circumference 48 cm (3rd–10th percentile). No dysmorphic features were observed (Fig.1). He presented severe axial hypotonia. He could hold eye contact only briefly before drifting away and he had no verbal communication.

Results of the WES Study
DNA Sample was sequenced with mean coverage of 109; 95% of target bases were covered at a minimum of 20×; 98% had coverage of minimum 10×. We filtered the results to retain high quality variants changing protein coding sequence or affecting splice sites with population frequency <0.01 (according to the EXAC database and an in house database of ∼500 Polish exomes). After filtering there were 254 variants left. These variants were searched for biallelic mutations consistent with autosomal recessive inheritance (Table I, Potentially AR) as well as for monoallelic variants potentially causative of an autosomal dominant or sex-linked condition (here, we considered variants not found in available databases and predicted to cause loss of protein function, i.e., introduce premature stop codon or affect a splice site (Table I, Potentially AD).
| Gene | Position | ID | Effect | Disease (inheritance) |
|---|---|---|---|---|
| Potentially AR (potentially biallelic variants, freq. <0.01 in available databases) | ||||
| ABCC1 | chr1:210004344-G>A | rs200922662 | NM_014388.6:p.Ser115Asn | Immunological phenotype in knock-out mice |
| chr16:016225712-C>T | rs200760311 | NM_004996.3:p.Arg1296Trp | ||
| TRIOBP | chr22:038134709-C>A | rs370224413 | NM_001039141.2:p.Gln1723Lys | Deafness, autosomal recessive 28; OMIM 609823 |
| chr22:038120377-G>A | rs202038718 | NM_001039141.2:p.Arg605Gln | ||
| HIC2 | chr22:021800637-G>A | − | NM_015094.2:p.Glu485Lys | − |
| chr22:021800187-C>T | NM_015094.2:p.Arg335Trp | |||
| DIEXF | chr1:210012290-C>T | − | NM_014388.6:p.Arg367Trp | − |
| chr1:210004344-G>A | NM_014388.6:p.Ser115Asn | |||
| Potentially AD (potential loss of function, freq. = 0 in available databases) | ||||
| GRIN2B | chr12:013724899-C>T | − | NM_000834.3:c.2011-1G>A | Epileptic encephalopathy, early infantile 27; OMIM 616139 |
| TUB | chr11:008122047-C>T | − | NM_003320.4:c.1282-3C>T | Retinal dystrophy and obesity, OMIM 616188 |
| DNAAF2 | chr14:050100715-C>A | − | NM_001083908.1:p.Glu385* | Ciliary dyskinesia, primary, 10, OMIM 612518 |
| NEK7 | chr1:198201761-AC>A | − | NM_133494.2:p.Pro18fs | − |
| SERINC3 | chr20:043132615-AG>A | − | NM_006811.2:p.Leu299fs | − |
- *Stop codon.
Analysis of available data on each of the genes harboring the variants after filtration (Table I) lead us to prioritize the heterozygous mutation in the last nucleotide of intron 10 of the GRIN2B gene (chr12:13724899 C>T, NM_000834.3:c.2011-1G>A). The identified mutation was confirmed by standard Sanger sequencing. Both parents were tested and no nucleotide changes were observed indicating de novo origin of the demonstrated mutation (Fig. 2). Paternity was verified using a panel of forensic STRs yielding odds >1,000,000:1 in favor of paternity. The WES study did not reveal other variants in any other potentially interesting genes.

In silico analysis using splice site prediction program confirmed that chr12:13724899 C>T disrupts the splice acceptor site, likely resulting in altered splicing.
DISCUSSION
We found a de novo GRIN2B mutation affecting a Guanine (G) at the 3′ end of the 9th intron. The G at this position is a constant feature in all major class introns, whereas in minor class introns it occurs alternatively with C but not other nucleotides [Pagani and Baralle, 2004]. Mutation of the −1 canonical splice site, together with the de novo occurrence (with maternity and paternity confirmed), strongly supports pathogenicity of a variant [Richards et al., 2015]. Furthermore, the pathogenicity of the c.2011-1G>A is consistent with its low frequency; the variant has not been previously reported in any of the available databases including the Exome Aggregation Consortium (Exac) database with 60,706 subjects (http://exac.broadinstitute.org).
Mutations in GRIN2A and GRIN2B, and chromosomal aberrations disrupting these genes, were described in patients with epilepsy with different severities accompanied by moderate to severe intellectual disability, but without defects visible in brain NMR and without specific dysmorphism [Endele et al., 2010; Epi4K Consortium et al., 2013]. At present no treatment is available, although Pierson et al. [2014] suggested that memantine (an NMDA receptor antagonist, until now used mainly in Alzheimer's, and never used as an anti-epileptic drug) may be effective in patients with a gain-of-function GRIN2A mutation. de Ligt et al. [2012] found missense de novo GRIN2B mutations in patients with intellectual disability by using diagnostic exome sequencing. Endele et al. [2010] discovered GRIN2B mutations in a large cohort of individuals with idiopathic epilepsy and/or intellectual disability. The majority of GRIN2B mutations in patients with epilepsy and epileptic encephalopathy described so far were missense mutations probably resulting in gain of function [Lemke et al., 2014]. Our data add to evidence that some loss of function GRIN2B mutations may also be a cause of epilepsy and epileptic encephalopathy.
Splice site GRIN2B mutations were previously described in patients with intellectual disability and/or autism spectrum disorders [Lemke et al., 2013, 2014; Hamdan et al., 2014; Kenny et al., 2014]. Interestingly, in a patient with West syndrome, Lemke et al. [2014] found a mutation (c.2011-5_2011-4delTC) located in the region of c.2011-1G>A which likely affected the very same splice site. Although c.2011-5_2011-4delTC was inherited, it is intriguing that it was located close to three other variants which were considered pathogenic due to de novo occurrence [Lemke et al., 2014]. As the authors discuss, the c.2011-5_2011-4delTC mutation could have a reduced penetrance similar to certain variants of GRIN2A causing idiopathic focal epilepsy with rolandic spikes [Lemke et al., 2013].
CONCLUSION
We identified an individual with severe intellectual disability and behavioral anomalies caused by early-onset epilepsy who had a heterozygous de novo GRIN2B splice-site mutation. This observation, together with a previous report [Lemke et al., 2014], suggests that epilepsy and early-onset EE may be caused not only by gain-of-function variants but also by splice site mutations—in particular those affecting the splice acceptor site of the 9th intron of the GRIN2B gene.
AUTHORS’ CONTRIBUTIONS
RS studied the patients, collected the samples, conceived the manuscript, supervised whole work. MB studied the patients, collected the samples, and helped in the clinical work. GK, JK, ES, AP, PK performed the experimental work. PS performed bioinformatics processing of NGS raw data. MS and KS critically revised the manuscript. RP conceived the study, supervised experimental work, analyzed the processed NGS data, and critically revised the manuscript.
ACKNOWLEDGMENTS
We are grateful to the patient and family members for participating in this study.