

Multiple capillary skin malformations, epilepsy, microcephaly, mental retardation, hypoplasia of the distal phalanges: Report of a new case and further delineation of a new syndrome†
How to Cite this Article: Isidor B, Barbarot S, Bénéteau C, Le Caignec C, David A. 2011. Multiple capillary skin malformations, epilepsy, microcephaly, mental retardation, hypoplasia of the distal phalanges: Report of a new case and further delineation of a new syndrome. Am J Med Genet Part A 155:1458–1460.
To the Editor:
Recently, Carter et al. [2001] reported two unrelated boys with multiple small capillary malformations on the skin from birth, intractable seizures, microcephaly, severe developmental delay, dysmorphic facial features, and hypoplasia of the distal phalanges. This association had never been reported before and was therefore considered as a new syndrome. Here, we report a third patient with striking phenotypic similarities. This paper further delineates this syndrome and confirms that it constitutes a distinct entity.
The propositus was the first child of healthy nonconsanguineous parents. Except for moderate maternal hypertension and gestational diabetes, pregnancy was uneventful. At term, the weight, length and occipitofrontal circumference (OFC) of the girl were 3,990 g (+1.8 SD), 49 cm (mean) and 32.5 cm (−1.8 SD), respectively. She presented with neonatal feeding difficulties but which did not require enteral nutrition. Physical examination showed a large anterior fontanel, anteverted nostrils, thin vermillion of the upper lip, abnormal auricles, short fifth fingers with hypoplastic nails of the second and fifth fingers, and short toes. Multiple small capillary malformations were present on the trunk, abdomen and limbs (Fig. 1). Neurological examination showed moderate axial hypotonia.

Multiple small capillary malformations on the trunk. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
At age 12 months, hospitalization in intensive care unit was required for a long lasting (>30 min) febrile hemiclonic seizures secondary generalized. Several episodes of seizure occurred during 48 hr despite treatment with Diazepam and Clonazepam. Valproic acid was started. Laboratory testing (electrolytes, calcium, blood gases) and cerebral MRI were normal.
At age 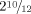 years, weight, height and OFC were 15.7 kg (+2 SD), 93 cm (mean), 47.5 cm (−2 SD), respectively. Walking was acquired at 22 months. Speech delay was noted as she was able to pronounce a few words from 18 months of age and to associate two words soon after. Her parents also noted aggressive behavior when frustrated. On examination she had a low frontal hairline, flat nasal ridge, flat nose, hypertelorism, bilateral epicanthus, thin upper lip, interrupted eyebrows (Fig. 2). Limbs examination showed short fingers with hypoplastic nails (Fig. 3), hypoplastic toes with clinodactyly of the second and fifth toes and pachyonychia on the fourth toes (Fig. 4). More than 30 small capillary malformations were still present and appeared to grow in proportion of the body size. Intercritic EEG was considered as normal and valproic acid was stopped at the age 5 years old.
years, weight, height and OFC were 15.7 kg (+2 SD), 93 cm (mean), 47.5 cm (−2 SD), respectively. Walking was acquired at 22 months. Speech delay was noted as she was able to pronounce a few words from 18 months of age and to associate two words soon after. Her parents also noted aggressive behavior when frustrated. On examination she had a low frontal hairline, flat nasal ridge, flat nose, hypertelorism, bilateral epicanthus, thin upper lip, interrupted eyebrows (Fig. 2). Limbs examination showed short fingers with hypoplastic nails (Fig. 3), hypoplastic toes with clinodactyly of the second and fifth toes and pachyonychia on the fourth toes (Fig. 4). More than 30 small capillary malformations were still present and appeared to grow in proportion of the body size. Intercritic EEG was considered as normal and valproic acid was stopped at the age 5 years old.

Facial appearance of the patient with low frontal hairline, flat nasal ridge, flat nose, bilateral epicanthus, hypertelorism, thin upper lip, and interrupted eyebrows. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]

Hands at age 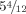 . Hypoplastic fifth fingers and hypoplastic nails. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
. Hypoplastic fifth fingers and hypoplastic nails. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]

Feet at age 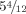 . Note irregular hypoplastic toes and nails. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
. Note irregular hypoplastic toes and nails. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
At last evaluation, age 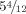 , her OFC was 47.5 cm (−2.5 SD). She was hospitalized for febrile status epilepticus which resolved after intravenous Phenobarbital, Clonazepam, and Phenytoin administration. Sodium valproate, which had been stopped 6 months earlier, was restarted. She was able to understand simple orders, to speak with short sentences and to write her name. Physical examination was unchanged. Karyotyping and array CGH (Agilent Human Genome CGH 44 K oligonucleotide arrays) were normal.
, her OFC was 47.5 cm (−2.5 SD). She was hospitalized for febrile status epilepticus which resolved after intravenous Phenobarbital, Clonazepam, and Phenytoin administration. Sodium valproate, which had been stopped 6 months earlier, was restarted. She was able to understand simple orders, to speak with short sentences and to write her name. Physical examination was unchanged. Karyotyping and array CGH (Agilent Human Genome CGH 44 K oligonucleotide arrays) were normal.
The patient reported here shares most of the features of the two unrelated patients described by Carter et al. [2001]. We can now distinguish a common phenotype shared by three patients including small capillary malformations on the skin from birth, delayed psychomotor development, epilepsy, microcephaly, and hypoplastic toes.
Small capillary malformations are one of the main clinical features of this syndrome. All three patients showed small capillary malformation evolving from birth. They are distributed all over the skin surface and are numerous (more than 30 in our patient). Clinical aspect and number do not evolve from birth, and they seem to grow with the body size. Psychomotor retardation ranges from moderate to severe. The first two patients described by Carter et al. had severe delay but our patient showed moderate mental retardation. Epilepsy was also severe in the first two patients who showed intractable seizures. It was less severe in our patient but she also showed one episode of status epilepticus. Microcephaly, progressive from birth ranging from −2.5 SD to −4 SD also seemed to be a constant finding. In the first two patients, cerebral MRI showed cortical atrophy and progressive thinning of the corpus callosum. These features were absent on our patient's MRI which could be due to a milder phenotype or to her relative young age when performing this exam (i.e., 12 months old). Our patient also shares some facial dysmorphic features with the two previous patients. Even though distal limb malformations are not specific to this syndrome, the morphological changes of the toes are very unusual. Toes are irregularly hypoplastic with clinodactylies and hypoplastic nails.
The two first reported patients were males. As our patient is female and all cases sporadic with nonconsanguineous parents, the most likely mode of inheritance is autosomal dominant due to de novo mutation. However, an X-linked trait or complex inheritance cannot be excluded.
Very few differential diagnoses could be discussed. So far, no syndrome has been reported with the association of developmental delay/mental retardation, capillary malformations, microcephaly and hypoplastic toes. In their article, Carter et al. discuss two other reports [Upton and Young, 1993; Leech et al., 2004] but which can be easily distinguished. In Upton and Young report, the patient showed skin lesions resembling hemangioma and there were no limb malformations. Regarding the patient reported by Leech et al. [2004], he had megalencephaly rather than microcephaly, and no limb abnormalities were noted.
To date, the molecular studies performed did not lead to the identification of the disease-causing gene. Microarray analyses were normal except for patient 1 [Carter et al., 2001] who carried a 307 kbp deletion inherited from his healthy mother. Direct sequencing of the two known deleted genes, GJB6 and CYL1, did not reveal any mutation. Due to the dermatological similarities with capillary malformation-arteriovenous syndrome (OMIM 608354), direct sequencing of the coding regions of the RASA1 gene was performed for the Patient 1 described by Carter et al. but failed to identify any mutation. Therefore, the pathophysiology leading to the phenotype of these patients remains unclear at this stage. Carter et al. posited that a gene involved in both vasculogenesis of the skin and distal limbs and neural survival may be responsible for this syndrome. There is the chance that new generation sequencing will lead to the identification of the disease-causing gene that hopefully will provide understanding of the mechanism leading to this disorder.
Acknowledgements
We are grateful to the family for their cooperation.

